




Occult hepatitis B virus infection (OBI) is characterized by the presence of replication-competent hepatitis B virus (HBV) DNA in the liver and/or serum of patients with undetectable levels of the HBV surface antigen (HBsAg). Due to the shared infection routes HIV positive patients are at higher risk of developing OBI, thus, the aim of this study was to determine the frequency of OBI in Mexican HIV-infected patients and to identify mutations in the HBV S gene that could be associated to the development of OBI.
Materials and methodsPlasma samples from 50 HIV-infected patients with undetectable levels of the HBsAg were obtained and analyzed. The Core, PreS and S genes were amplified by nested PCR and sequenced by the Sanger method. To analyze HBV diversity in the OBI-positive patients, ten sequences of 762bp from the HBV S gene were selected, cloned, and subsequently sequenced for mutational analyses.
ResultsOBI infection was found with a frequency of 36% (18/50). All the HBV sequences corresponded to the H genotype. The most common mutations were: C19Y, Q129H, E164D, and I195M, with a frequency of 44%, 36%, 39% and 48% respectively.
ConclusionsIn this study, we report the presence of OBI in a cohort of Mexican HIV-infected patients with an overall prevalence of 36%. Mutational analyses revealed that four non-silent mutations were frequent in different regions of the HBsAg gene, suggesting that they might be associated to the development of OBI in this population, nevertheless, further studies are required to determine their role in the pathogenesis of OBI.
Hepatitis B virus (HBV) infections are considered an important public health issue, particularly due to their endemicity in many countries in the world. Between 350 and 400 million people worldwide are infected with HBV and show signs of chronic HBV infection. The occurrence of coinfections between human immunodeficiency virus (HIV) and HBV is very common, mainly because of the similar infection routes that both viruses share, and according to the Global Hepatitis Report 2017 (World Health Organization), the prevalence of HBV infection in HIV-infected persons is 7.4% [1].
The main serological marker for the detection of HBV is the surface antigen (HBsAg) and the persistence of this antigen in the blood of infected patients for more than six months, defines the chronic phase of HBV infection the disease. In contrast, occult hepatitis B virus infection (OBI) is defined by undetectable levels of the HBsAg or anti-HBsAg antibodies in the blood of infected individuals accompanied by the presence of either, episomal HBV covalently closed circular DNA (cccDNA) in the liver, or detectable levels of HBV DNA in the blood [2].
There is a clear correlation with the prevalence of OBI and the level of endemicity of HBV in different geographic regions. Interestingly, highly variable levels of OBI prevalence, ranging between 0 and 90% have been reported, depending on the HBV genotype and the method used for the detection [3–5]. It is worth mentioning that HIV positive patients show a higher risk of developing OBI [4,6].
In Mexico, despite the low prevalence of chronic HBV infection (0.21%) and the high level of vaccine coverage (92%) [7–9], mandatory in the national vaccination scheme since 1999, two studies regarding the presence of OBI in HIV positive patients have been published. The first study by Torres-Baranda et al. reported an OBI prevalence of 18.4% in HIV-positive patients using only one molecular marker (S gene) and a nested PCR assay designed to detect the DNA-HBV in serum samples [10]. In the second study, we reported an OBI frequency of 49% using two out of three positive molecular markers as a criteria to detect OBI and a nested PCR to amplify the Core region and two real time PCR assays to amplify the X and S genes in plasma samples [11].
The molecular mechanisms associated to the development of OBI are still unknown, nevertheless, several hypotheses have been proposed to explain this phenomenon. Previous studies have suggested that mutations in the PreS/S ORF that codes for the HBsAg might be related to the lack of detection of the HBsAg in the blood of infected patients, possibly due to antigenic changes that reduce the affinity of diagnostic antibodies toward this protein or to the reduced secretion of the HBsAg into the bloodstream [12,13]. To date, few studies have reported the molecular features of OBI in Mexico. In this study, we describe the frequency and the mutations in the S gene of occult HBV in patients with HIV.
2Materials and methods2.1Patient SamplesPlasma samples were collected from 50 HIV-infected patients attending the Hepatitis Clinic from the Infectology Hospital CMN “La Raza”, Instituto Mexicano del Seguro Social (IMSS). All the patients enrolled were adults and showed undetectable levels of the HBsAg, nevertheless, the presence of antibodies against HBsAg as a serological marker of previous exposure to HBV was not considered as an exclusion criterion. Every patient enrolled in the study signed informed consent forms. This project was approved by the Institutional Ethics Committee with registration number R-2018-785-009.
2.2DNA extraction and detection of HBV-DNADNA was extracted from 100μL of plasma using the QIAamp DNA Mini Kit (QIAGEN GmbH, Hilden, Germany) following the manufacturer's instructions. Detection of HBV was carried out using nested PCR assays with the Core, PreS and S genes as follows.
2.3Nested PCR assays for the HBV Core, PreS and S genes.A 560bp fragment of the Core gene was amplified with the following primers: Fwd_HBOC-1 (5′-TTCAAGCCTCCAAGCTGTGCCTTGG-3′, nt 1863–1887) and Rev_HBOC-2 (5′-TCTGCGACGCGGCGATTGAGA-3′, nt 2402–2422). Subsequently, 438bp fragments were amplified by nested PCR with the following primers: Fwd_HBOC-3 (5′-CCTTGGGTGGCTTTGGGGCA-3′, nt 1882–1901) and Rev_HBOC-4 (5′-AGGATAGGGGCATTTGGTGGTCTATA-3′, nt 2294–2319) [11].
Primer pairs to amplify fragments of the hepatitis B genome genotype H covering the PreS and S regions were designed with the Primer CLC Main Workbench version 6.6.1 software (CLCBio, Aarhuf, Denmark). The conserved sites in these regions were determined by multiple sequence alignments against the set of the three HBV genotype H reference sequences (Accession numbers: AY090454.1, AY090457.1, AY090460.1).
A 741bp fragment of the PreS gene was amplified with the following primers: Fwd_PreSF-EXT (5′-ATACTTCCCTTTGGATAAAGG-3′ nt 2671–2692), and Rev_PreSR-INT (5′-CCTGATGTGATGTTCTCCAT-3′ nt 178–197), subsequently a nested PCR was used to amplify a 630bp fragment using the Fwd_PreSF-INT (5′-ATTTACATACTTTGTGGAAGGC-3′ nt 2748–2770) and Rev_PreSR-EXT (5′-TGTGATGTTCTCCATGTTCAT-3′ nt 151–172), primers.
Finally, a 953bp fragment from the S gene was amplified using the following primers: Fwd_HBV1-F (5′-GCTGGTGGCTCCAGTTCAGAAACACAGA-3′ nt 59–86) and Rev_ HBV2-R (5′-CAAAAGACCCACAATGCGTTGACAAACC-3′ nt 984–1011). Next, a 762bp fragment was amplified by nested PCR using the primers Fwd_PolRT-3F (5′-CGAAGACTGGGGACCCTGC-3′, nt 129–147) and Rev_HBO2-R (5′-ATATAACCCATAAAGTGTAAGGAATA-3′ nt 864–890). For the first amplification step 1μL of DNA was added to the reaction mixture, for the nested PCR we added 1μL of the first round product. Every PCR assay was carried out in a final volume of 12.5μL with the following reaction mixture: 0.3U of Ex Taq HS TAKARA (Takara Bio USA, Inc.), 1× Ex Taq Buffer that contains 2mM MgCl2, 0.2mM dNTPs (Takara Bio USA, Inc.) and 0.2μM of each primer.
For the accurate detection of the HBV-DNA in patients with OBI, a LOD of 100copies/mL for each nested PCR reaction used to detect the three molecular markers are needed. Thus, we evaluated the sensitivity of each PCR assay using serial tenfold dilutions (1:10 to 1:10,000) of an HBV DNA sample (HBV-133) with a viral load of 270,000copies/mL, previously quantified using the Cobas Taqman (CTM) HBV test (Roche, Diagnostics, Germany).
Each assay included a DNA sample from a patient infected with HBV (viral load: 840,000copies/mL) as a positive control and DNA extracted from a sample with negative serologic markers and undetectable viral load for HBV as a negative control. A negative control of dH2O was included. The samples were run in triplicate and each test was repeated at least two times. Extreme care was taken to avoid cross-contamination between samples. In addition, all the experimental work regarding sample and PCR product manipulation was carried out in specific laboratory work areas were designated for handling of sample and for the manipulation of PCR products. PCR products were analyzed in 1% agarose gels stained with HydraGreen™ (ACT Gene, Piscataway, NJ).
To confirm the negativity of the samples, a second DNA extraction was carried out using the High Pure Viral Nucleic Acid Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions and the nested PCRs were repeated as previously described.
2.4Sanger sequencingPCR products were purified by ethanol precipitation and resuspended in dH2O before Nanodrop quantification. 10ng of the purified PCR fragments were sequenced using the Big Dye® Terminator v3.1 kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer's instructions. All the sequences were obtained using the Applied Biosystems 3730xl DNA Analyzer.
2.5Cloning and sequencing of the HBV S geneFor cloning, ten amplicons corresponding to the 762bp fragment of the S gene from HBV patients with OBI were selected. These products were purified by ethanol precipitation and cloned into the pJET1.2/blunt vector (Clone JET™ PCR Cloning Kit Thermo Scientific Inc) according to the manufacturer's instructions.
The recombinant plasmids were transformed into Escherichia coli JM-109 and ten clones per sample were selected and amplified in liquid Luria Bertani (LB) medium with 100μg/mL of ampicillin. Ten clones from each sample were randomly selected and plasmid extraction was carried out from each of the selected clones, using the FavorPrep™ Plasmid Extraction Mini Kit (FAVORGEN Biotech Corp, Taiwan). The presence of the inserts was verified by PCR and then 80ng of the positive plasmids were subsequently sequenced using the Big Dye® Terminator v3.1 kit (Applied Biosystems, Foster City, CA, USA), using the universal primers pJET1.2 Forward nt 310–332 y pJET1.2 reverse nt 428–405.
2.6GenotypingFor the genotypification of HBV, the S sequences obtained from OBI patients were analyzed using the Genotyping tool available at the National Center for Biotechnological Information (NCBI) website (https://www.ncbi.nlm.nih.gov/projects/genotyping/formpage.cgi). The 18 S sequences were aligned against the 23 sequences of the S gene representative of the eight HBV genotypes available at the NCBI genotyping tool. A phylogenetic tree was built using the test of Maxium Likelihood and with a bootstrap of 1000 replicates using MEGA 6.06 software.
2.7Mutational analysis of the S gene from HBVTo identify mutations in the S gene related to the presence of OBI, the nucleotide sequences that were cloned and later sequenced were in silico translated using the MEGA 6.06 software. The criteria to define a mutation site in the amino acid sequence was the presence of changes in S gene that were not present in the set of three reference sequences belonging to genotype H, available at the NCBI genotyping tool. Then, the frequency of all the non-synonymous mutations identified their frequency per clone and per sample. Additional mutational analyses were carried out using 98 deduced amino acid sequences of the S gene from the HBVdb (https://hbvdb.lyon.inserm.fr/HBVdb/HBVdbDataset?view=/data/proteins/fasta/H_SHBs.fas&seqtype=2) and 17 sequences obtained from the Hepatitis Virus Diversity Research Unit (HVDR) database [14] (http://hvdr.bioinf.wits.ac.za/alignments/index.html). Multiple sequence alignments were built using the Clustal W method with MEGA 6 software.
3Results3.1Patient InformationSociodemographic data was collected from the 50 HIV-infected patients included in the study 76% (38/50) were men, with a median age of 48 years, the viral load of HIV-RNA was undetectable in 74% of the patients (34/50), in 26% of patients, the mean of HIV-RNA VL log10 copies/mL was 2.6. The average count of CD4+ was of 578 (Table 1).
Demographic and clinical data of HIV patients.
| Patients HIV-1 infected negative HBsAg | n=50 |
| Gender M:F | 38:12 |
| Age range (yrs) | 31–66 |
| Range of HIV RNA viral load in plasma (log10copies/mL) | NDa-5.1 |
| CD4+ count cells/μL mean | 578 |
| AST values mean (U/L) | 27.6±7.6 |
| ALT values mean (U/L) | 36±20 |
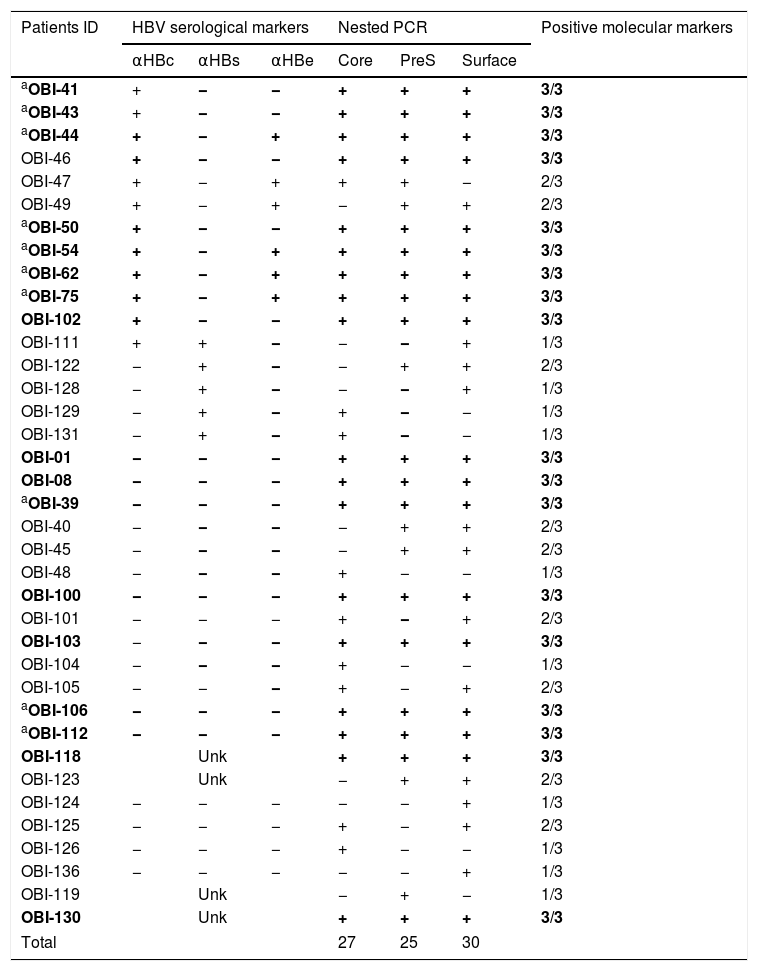
HBV-DNA was detected in 74% of the analyzed samples (37/50), the rest (26%) were negative. Of the positive samples, 10 were positive for one genomic region, nine were positive for two, and 18 were positive for the three genomic regions (Table 2), meeting the criteria to be considered as OBI [2]. In this group of HIV-infected patients, we found that the frequency of OBI was 36% (18/50). The sequence of the PCR products for the confirmed OBI cases was determined and the genotypification analyses carried out with the NCBI Genotyping tool indicated that all the sequences belonged to genotype H (data not shown).
Detection of hepatitis B virus DNA regions, serologic markers in HIV-infected patients with negative HBsAg.
| Patients ID | HBV serological markers | Nested PCR | Positive molecular markers | ||||
|---|---|---|---|---|---|---|---|
| αHBc | αHBs | αHBe | Core | PreS | Surface | ||
| aOBI-41 | + | − | − | + | + | + | 3/3 |
| aOBI-43 | + | − | − | + | + | + | 3/3 |
| aOBI-44 | + | − | + | + | + | + | 3/3 |
| OBI-46 | + | − | − | + | + | + | 3/3 |
| OBI-47 | + | − | + | + | + | − | 2/3 |
| OBI-49 | + | − | + | − | + | + | 2/3 |
| aOBI-50 | + | − | − | + | + | + | 3/3 |
| aOBI-54 | + | − | + | + | + | + | 3/3 |
| aOBI-62 | + | − | + | + | + | + | 3/3 |
| aOBI-75 | + | − | + | + | + | + | 3/3 |
| OBI-102 | + | − | − | + | + | + | 3/3 |
| OBI-111 | + | + | − | − | − | + | 1/3 |
| OBI-122 | − | + | − | − | + | + | 2/3 |
| OBI-128 | − | + | − | − | − | + | 1/3 |
| OBI-129 | − | + | − | + | − | − | 1/3 |
| OBI-131 | − | + | − | + | − | − | 1/3 |
| OBI-01 | − | − | − | + | + | + | 3/3 |
| OBI-08 | − | − | − | + | + | + | 3/3 |
| aOBI-39 | − | − | − | + | + | + | 3/3 |
| OBI-40 | − | − | − | − | + | + | 2/3 |
| OBI-45 | − | − | − | − | + | + | 2/3 |
| OBI-48 | − | − | − | + | − | − | 1/3 |
| OBI-100 | − | − | − | + | + | + | 3/3 |
| OBI-101 | − | − | − | + | − | + | 2/3 |
| OBI-103 | − | − | − | + | + | + | 3/3 |
| OBI-104 | − | − | − | + | − | − | 1/3 |
| OBI-105 | − | − | − | + | − | + | 2/3 |
| aOBI-106 | − | − | − | + | + | + | 3/3 |
| aOBI-112 | − | − | − | + | + | + | 3/3 |
| OBI-118 | Unk | + | + | + | 3/3 | ||
| OBI-123 | Unk | − | + | + | 2/3 | ||
| OBI-124 | − | − | − | − | − | + | 1/3 |
| OBI-125 | − | − | − | + | − | + | 2/3 |
| OBI-126 | − | − | − | + | − | − | 1/3 |
| OBI-136 | − | − | − | − | − | + | 1/3 |
| OBI-119 | Unk | − | + | − | 1/3 | ||
| OBI-130 | Unk | + | + | + | 3/3 | ||
| Total | 27 | 25 | 30 | ||||
αHBc, HBV core antibody; αHBs, HBV surface antibody; αHBe, HBV e antigen antibody; positive, +; negative, −; Unk, unknown.
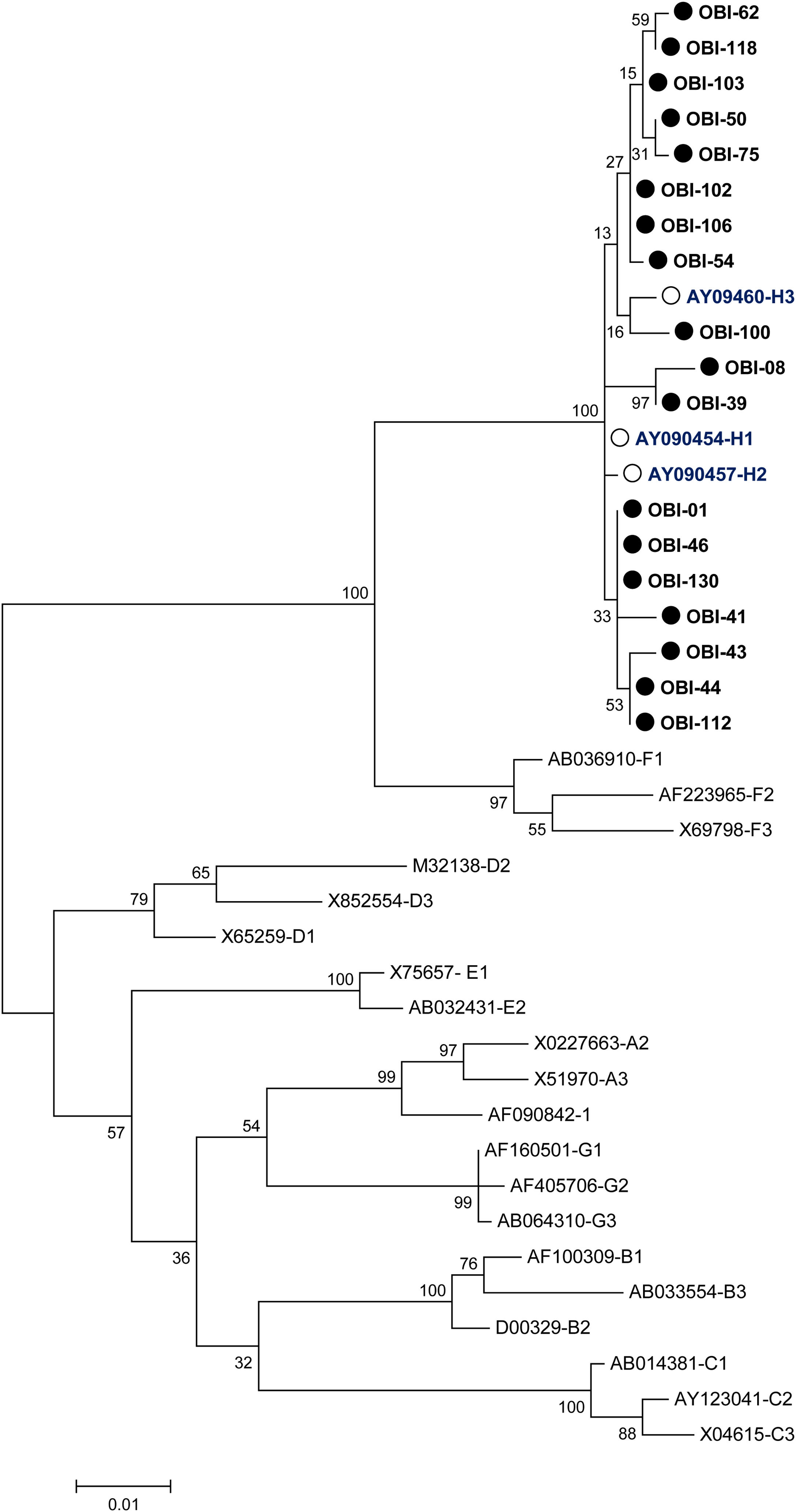
A phylogenetic tree (Fig. 1) was built with the 18 OBI S gene sequences and the reference sets of sequences from all the HBV genotypes (A-H) available at the HBV genotyping tool on the NCBI website, showing that all of the OBI sequences belonged to genotype H.
 isolates from 18 patients with occult HBV infection (filled circle) and including 23 GenBank sequences of genotypes A–H (GenBank accession number and HBV genotype indicated, genotype H unfilled circle). The bootstrap values are indicated for the major nodes as a percentage of the data obtained from 1000 replicates.")
Phylogenetic tree constructed by the Maximum Likelihood analysis of the S region of hepatitis B virus (HBV) isolates from 18 patients with occult HBV infection (filled circle) and including 23 GenBank sequences of genotypes A–H (GenBank accession number and HBV genotype indicated, genotype H unfilled circle). The bootstrap values are indicated for the major nodes as a percentage of the data obtained from 1000 replicates.
Nine of the 18 OBI patients were considered as seropositive (SP) because they had one or two HBV serological markers, HBV core antibody (αHBc) and HBV e Antigen antibody (αHBe), the other nine patients were negative for serological markers and were considered as seronegative (SN) for OBI (Table 2).
3.3Mutational analysis3.3.1Identification of mutations in OBI patientsTo identify mutations in the HBsAg, the nucleotide sequences of the S gene from the 18 confirmed OBI cases were aligned with three reference sequences belonging to genotype H of HBV (GenBank, NCBI). The amino acid sequence for the fragments was deducted with the CLC Main Workbench and afterwards, consensus sequences were determined.
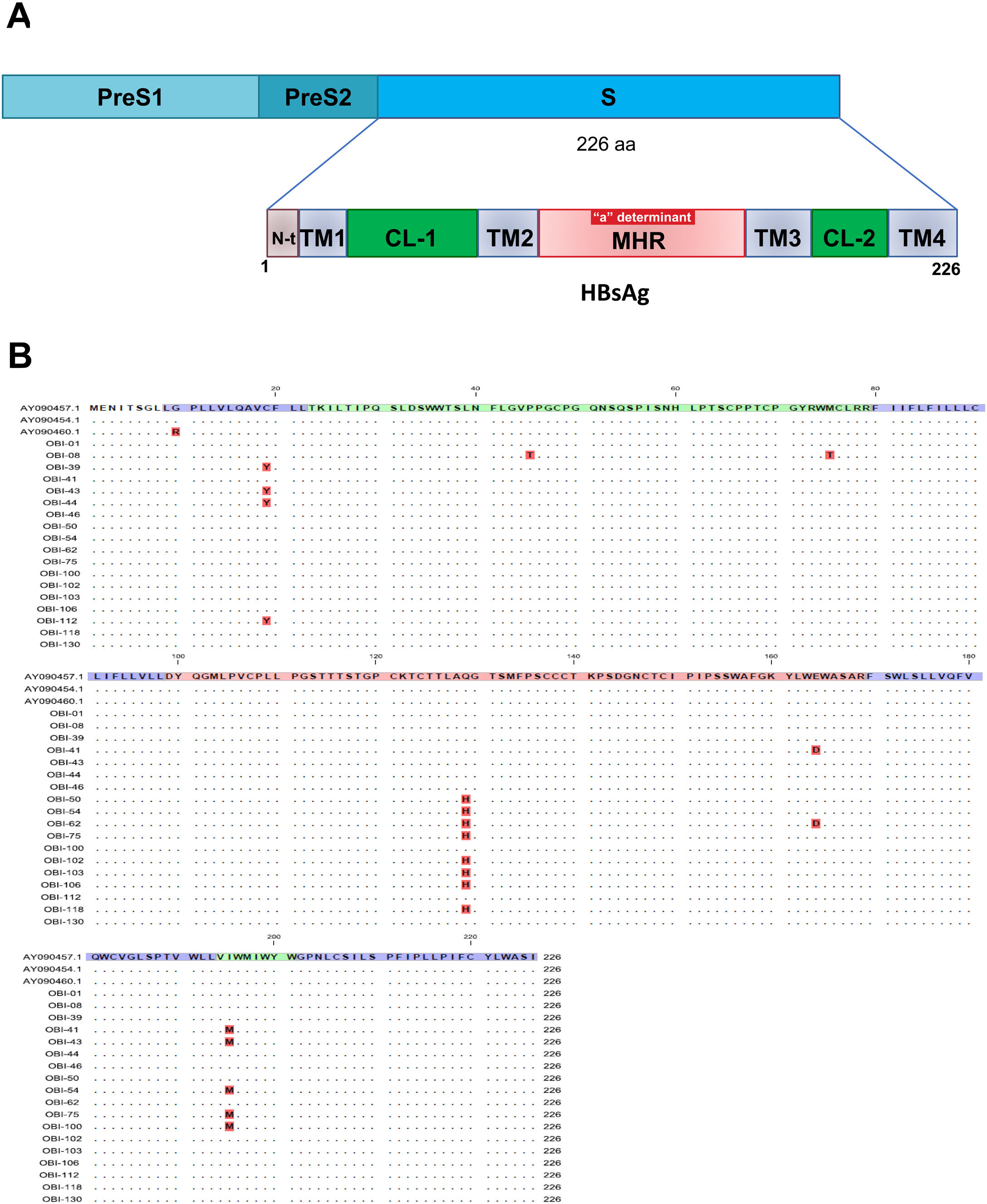
Mutations were identified according to their position in the different regions of the HBsAg, which is constituted by a first transmembrane domain (TM1) which goes from residues 8 to 23, the cytosolic loop (CL-1) from residues 24–79. The second transmembrane domain (TM2) from residues 80–98, the major hydrophilic region (MHR) from residues 99–169. The third transmembrane domain (TM3) from residues 170–193, the second cytosolic loop (CL-2) from residues 194–201 and finally the fourth transmembrane domain (TM4) from residues 202–226 (Fig. 2A). The most frequent mutations observed were: Q129H with 44% (8/18), followed by I195M with 28% (5/18), C19Y with 22% (4/18) and E164D with 11% (2/18) (Fig. 2B).
 The ORF-S encoding the hepatitis B surface antigen is depicted. The lower schematic highlights functional regions of the HBsAg. TM1, transmembrane domain residues 8–23; the cytosolic loop (CL-1) residues 24–79; TM2 residues 80–98; MHR residues 99–169; TM3 residues 170–193; the second cytosolic loop (CL-2) residues 194–201 and the TM4 residues 202–226. B) Alignment of amino acid sequences deduced from OBI patients. Residues identical to the genotype H reference consensus are indicated by dots. Mutations are indicated in red boxes.")
Mutational profile of the HBsAg among 18 OBI patients genotype H. (A) The ORF-S encoding the hepatitis B surface antigen is depicted. The lower schematic highlights functional regions of the HBsAg. TM1, transmembrane domain residues 8–23; the cytosolic loop (CL-1) residues 24–79; TM2 residues 80–98; MHR residues 99–169; TM3 residues 170–193; the second cytosolic loop (CL-2) residues 194–201 and the TM4 residues 202–226. B) Alignment of amino acid sequences deduced from OBI patients. Residues identical to the genotype H reference consensus are indicated by dots. Mutations are indicated in red boxes.
Since a complex mixture of viral populations or quasispecies circulates in the plasma of infected patients, we decided to clone the S fragment from patients with OBI. Only 10 fragments from different samples were successfully cloned.
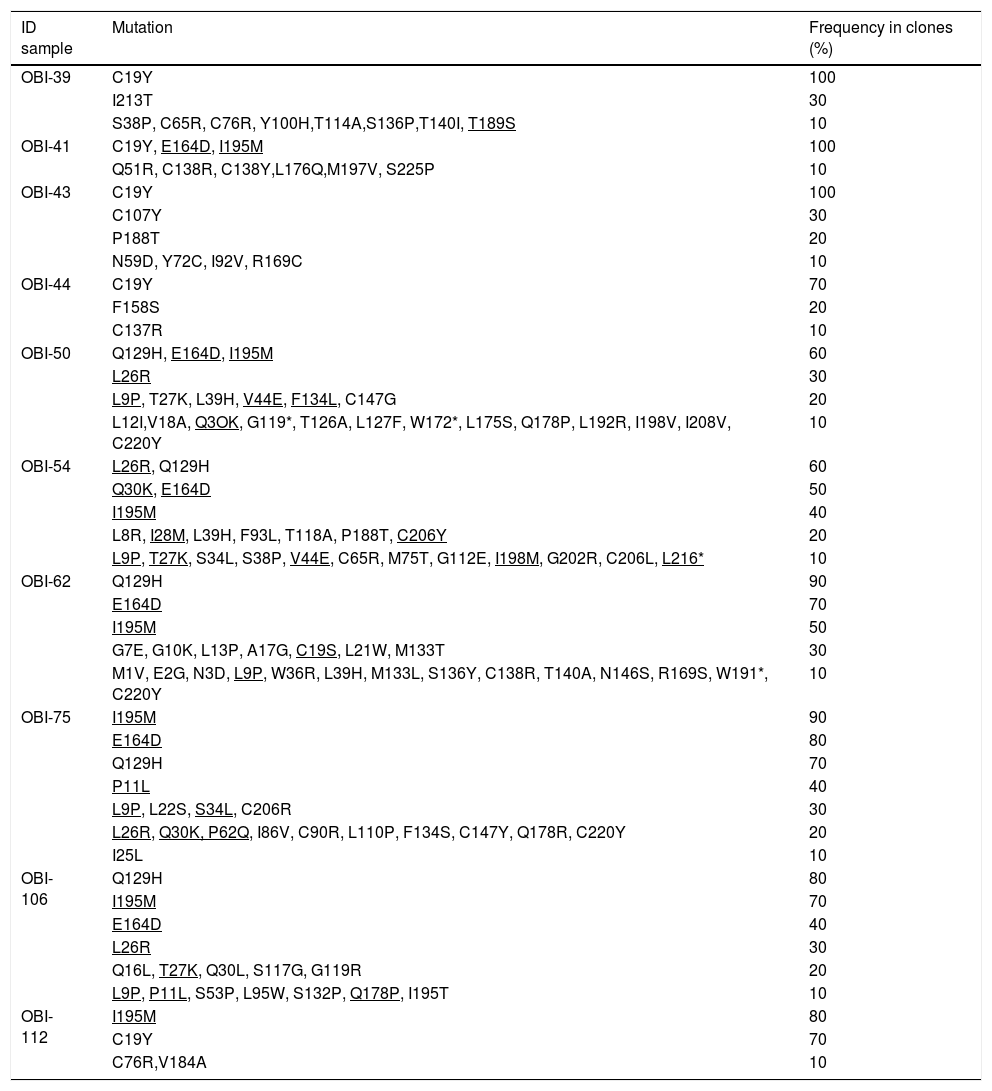
The clones per sample were selected by PCR and sequenced. The analysis of 100 sequences obtained from the clones was useful to study the genetic variability, even between clones derived from a single patient (Supplementary Fig. 1). In Table 3, all the HBsAg mutations identified per patient are described. The most frequent mutations were: C19Y (44%), Q129H (36%), E164D (39%) and I195M (48%).
Mutations in the HBsAg detected by clone-based sequencing.
| ID sample | Mutation | Frequency in clones (%) |
|---|---|---|
| OBI-39 | C19Y | 100 |
| I213T | 30 | |
| S38P, C65R, C76R, Y100H,T114A,S136P,T140I, T189S | 10 | |
| OBI-41 | C19Y, E164D, I195M | 100 |
| Q51R, C138R, C138Y,L176Q,M197V, S225P | 10 | |
| OBI-43 | C19Y | 100 |
| C107Y | 30 | |
| P188T | 20 | |
| N59D, Y72C, I92V, R169C | 10 | |
| OBI-44 | C19Y | 70 |
| F158S | 20 | |
| C137R | 10 | |
| OBI-50 | Q129H, E164D, I195M | 60 |
| L26R | 30 | |
| L9P, T27K, L39H, V44E, F134L, C147G | 20 | |
| L12I,V18A, Q3OK, G119*, T126A, L127F, W172*, L175S, Q178P, L192R, I198V, I208V, C220Y | 10 | |
| OBI-54 | L26R, Q129H | 60 |
| Q30K, E164D | 50 | |
| I195M | 40 | |
| L8R, I28M, L39H, F93L, T118A, P188T, C206Y | 20 | |
| L9P, T27K, S34L, S38P, V44E, C65R, M75T, G112E, I198M, G202R, C206L, L216* | 10 | |
| OBI-62 | Q129H | 90 |
| E164D | 70 | |
| I195M | 50 | |
| G7E, G10K, L13P, A17G, C19S, L21W, M133T | 30 | |
| M1V, E2G, N3D, L9P, W36R, L39H, M133L, S136Y, C138R, T140A, N146S, R169S, W191*, C220Y | 10 | |
| OBI-75 | I195M | 90 |
| E164D | 80 | |
| Q129H | 70 | |
| P11L | 40 | |
| L9P, L22S, S34L, C206R | 30 | |
| L26R, Q30K, P62Q, I86V, C90R, L110P, F134S, C147Y, Q178R, C220Y | 20 | |
| I25L | 10 | |
| OBI-106 | Q129H | 80 |
| I195M | 70 | |
| E164D | 40 | |
| L26R | 30 | |
| Q16L, T27K, Q30L, S117G, G119R | 20 | |
| L9P, P11L, S53P, L95W, S132P, Q178P, I195T | 10 | |
| OBI-112 | I195M | 80 |
| C19Y | 70 | |
| C76R,V184A | 10 |
The * symbol indicates a stop codon. Mutations that were identified also in other HBV sequences genotype H are underlined.
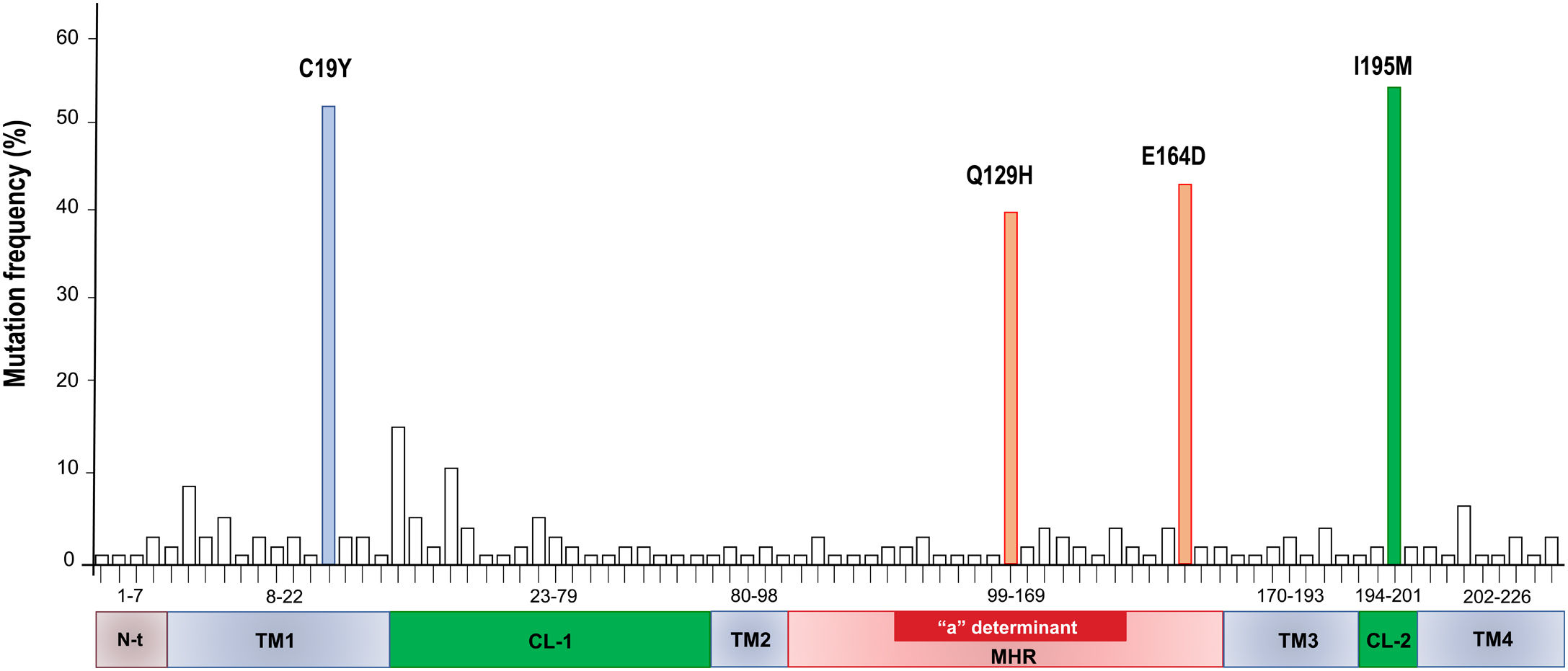
A total of 86 HBsAg mutations were identified in the different domains of the protein (Fig. 3). A frequency of 24% (12/86) of the mutations were found in the TM1, 23% (20/86) in the CL-1, 6% (5/86) in the TM2, 25.5% (22/86) in the MHR, 9% (8/86) in TM3, 3.5% (3/86) in the CL-2 and 10% (9/86) in TM4
4Discussion
In this study, we report a frequency of occult hepatitis B virus infections of 36% in a cohort of 50 Mexican patients infected with HIV. Previous reports worldwide have demonstrated that prevalence data is variable, ranging from 1 to 89% [4]. In Mexico, only two studies have reported the frequency of OBI in HIV positive patients. Torres R et al. reported a prevalence of 18.4% (7/38) in HIV patients from Jalisco, using as a criteria for OBI infection the positivity with only one molecular marker, the S-gene [10]. The second report, from our research group, reported a prevalence of 49% in HIV patients from Mexico City, using as a criteria the positivity of at least two of three molecular markers [11]. The differences reported in these two studies might be associated with several factors which include the level of endemicity, the inclusion criteria used in the studies, the size and type of sample, the sensitivity and specificity of the assays, as well as the sets of primers used for the detection of HBV genome [15–19].
The golden standard for the confirmation of OBI is the detection of HBV-DNA in serum or liver tissue samples, and requires assays that demonstrate to be highly specific and sensitive, due to the fact that HBV DNA in OBI patients is found in very low concentrations (<200IU/mL), moreover, previous studies have demonstrated that intermittent detection of HBV-DNA in OBI patients is not a rare event [2,20].
In this study, based on the Taormina criteria [2], we determined the frequency of occult hepatitis B virus infections in a group of HIV positive patients from a tertiary care hospital in Mexico City, using three standardized nested PCR assays to amplify the Core, PreS and S genomic regions of HBV genotype H. These assays showed a limit of detection of around 100copies/mL (approximately 20 UI/mL), which is consistent with previous reports that demonstrated that the viral load in OBI patient is about 20IU/mL [17]. In spite of there are several commercial kits to quantify the HBV viral load by quantitative PCR, such as the COBAS AmpliPrep-COBAS TaqMan 48, which has a lower detection limit of 10–20 UI/mL [21], to date, they have not been validated for the detection of OBI.
OBI infections are multifactorial entities and although several mechanisms for the development of OBI have been suggested, none of them have been fully demonstrated. Among them, we can mention co-infections with other viruses such as HCV and HIV, factors related to altered immune responses, genome integration of some regions of the HBV that could be associated with the absence of HBsAg and the low level of virus replication, and mutations in all the HBV genome regions [22,23].
Most of the mutational analysis of the HBsAg that have been carried out to search for possible associations between them and the development of OBI, nevertheless, they have only focused on the MHR region of the S gene, particularly in the “a” determinant, a highly conserved antibody-neutralizing epitope cluster [24–26]. Some of these mutations have been associated to the lack of detection of the HBsAg due to structural and antigenic changes in the protein, causing the common tests to throw back false negative results. Moreover, mutations in other sited of the HBsAg might be causing alterations in the maturation, secretion and expression of the HBsAg, highlighting the importance of analyzing the whole protein in search for mutations [27–29].
We aimed to determine if mutations in the HBsAg, in this cohort of Mexican patients also infected with HIV, might be associated with the development of OBI. Considering that during the HBV replication, genetic variants or viral quasispecies are generated [30], the S gene sequences from ten OBI patients were amplified and cloned. We did not search for specific mutations associated to OBI; our approach was to compare the S gene sequences obtained from OBI patients with the set of three reference sequences genotype H from the NCBI genotyping tool. Then, the nonsynonymous mutations were listed and the frequency among clones in one sample, as well among all the samples was determined.
The cloning strategy resulted to be useful to confirm that several quasispecies of HBV can be found in one single patient. These quasispecies are not identical but are related between them (see Supplementary Fig. 1, Suppl. Fig. 2, Table 3). Moreover, we identified several mutations in all the domains that constitute de ORF-S. A total of four non-synonymous mutations were identified: one in the TM1 domain (C19Y), two in the MHR (Q129H, E164D) and one in the CL-2 (I195M).
The C19Y mutation was found with a frequency of 44%, and it is located in a region that is necessary for the insertion and correct folding of the S protein during its translation in the endoplasmic reticulum [31]. If any of these mutations is present in the TM1 domain, it could have an effect on the secretion of the HBsAg and the HBV virions during replication cycle [32,33]. The C19F mutation was also reported in a Mexican patient from the Nahua genotype H with OBI by Roman et al.[34]. Even though this study was descriptive, it is interesting that this mutation was found in an HIV negative patient, suggesting that his mutations might also be present in other groups of patients with OBI. The substitution is F19V was reported in a group of OBI donors with HBV genotype C, nevertheless, since it was found in a different genotype, the amino acid substitution is different. The authors did not report any function or association with OBI [35].
The Q129H substitution located within the “a” determinant of HBsAg, was found in 36% of the OBI patients. This mutation, described as an escape mutation [36], has also been reported in other genotypes but mainly in blood donors [27,37–39]. This mutation was reported in 5/21 blood donors recently diagnosed with HBV in Mexico [40]. It is interesting that this escape mutant has not been reported in OBI patients with genotype H in Mexico. More studies are needed to understand the role of these mutations in the OBI mechanisms.
Other frequent mutation found in the MHR was E164D (39%). This mutation is often found as a double mutation along with I195M. These mutations alter the antigenicity of HBsAg and change its affinity for anti-HBsAg specific antibodies. Some of these mutations, for example, those corresponding to the I195M, W196S, and the M198I mutants, affect a conformational region of the HBsAg protein consisting of two short alpha helixes separated by a disulphide bridge forming a turn [41]. Besides, this substitution can produce simultaneous mutations in both, the overlapping surface antigen, and the polymerase ORF's. When is present as a double mutation (sE164D/sI195M), it has been associated with the triple lamivudine resistance polymerase mutation pattern (rtV173L, rtL180M, rtM204V).
We detected the double mutation, sE164D/sI195M, in six OBI patients. Of these, two (OBI-41, OBI-62) included lamivudine (LMV), and four patients (OBI-50, 54, 75,106) tenofovir in their ARV treatment for HIV infection.
One patient (OBI-112) had only the I195M mutation, which simultaneously produces the rtM204V, that induces resistance to LMV. Because this patient was naive to treatment, this drug resistance mutation could be a naturally induced genetic variant of HBV. This phenomena has been previously reported in an untreated Mexican patient with chronic HBV infection genotype H and coinfected with HIV, probably due to immune pressure as suggested by the author [42].
We found two patients (OBI-43 y 44) that were under treatment with LMV and did not show resistance-associated mutations. In this case, some studies have suggested that these mutations might be found in a minority of the HBV quasispecies from a patient (<20%), thus to detect these mutations, highly sensitive cloning methods are required [43]. Nevertheless, the cloning and direct sequencing method used in this study has a low sensitivity for the detection of underrepresented variants (<20%), thus, we cannot rule out some genetic variants of HBV that might be present in a single sample could have not been detected.
This study has some limitations: the number of patients included (50), it would be necessary to analyze more HIV positive patients to associate the presence of these mutations with the OBI infection, and the association with clinical and demographic variables. We were not able to clone eight OBI samples, probably due to some technical issues and in consequence, these OBI samples were not included in the mutational analysis. The other limitation is the number of clones analyzed, ten per sample. In other studies it has been suggested to analyze up 100 clones per sample, or to use of more sensitive methods such as high throughput sequencing and ultradeep pyrosequencing as an alternative to study the genetic variability of HBV in a single sample [43,44].
In the last years, research carried out to detect occult hepatitis B virus infections have demonstrated the consequences of this type of infections on public health systems worldwide. Detection of OBI is expensive and technically demanding, thus, many patients with OBI might remain undiagnosed. Besides, OBI can be transmitted by blood transfusion and organ transplants and is considered a risk factor for future development of chronic hepatic diseases such as hepatocellular carcinoma and cirrhosis.
5ConclusionIn this study, we report a frequency of occult hepatitis B virus infections of 36% in a cohort of Mexican HIV-infected patients highlighting the importance of using sensitive HBV-DNA detection techniques as a prophylactic measure in this type of highly susceptible population.
Several mutations, C19Y, Q129H, E164D, I195M, that were present with a high frequency were also identified and might be associated to the development of OBI. Nevertheless, there are still several questions to solve, and further studies are needed to evaluate the role and function of these mutations in the development of OBI.
AbbreviationsHBV hepatitis B virus human immunodeficiency virus hepatitis C virus occult hepatitis B virus infection hepatitis B surface antigen covalently closed circular DNA first transmembrane domain first cytosolic loop second transmembrane domain major hydrophilic region the second cytosolic loop the third transmembrane domain fourth transmembrane domain seropositive for HBV serological markers seronegative for HBV serological markers HBV surface antibody HBV core antibody HBV e antigen antibody lamivudine surface reverse transcriptase N-terminal
The authors declares that there is no conflict of interest regarding the publication of this article
FundingThis work was supported by the Instituto Mexicano del Seguro Social (IMSS) (FIS/IMSS/PROT/G18/1819).
Karina Enriquez Navarro was PhD scholarship holder from CONACYT (307535) in the program in Experimental Biology, DCBS, Universidad Autónoma Metropolitana Iztapalapa, Mexico, and a scholarship from the IMSS.
The following are the supplementary data to this article:
Deduced amino acid sequence analysis of the HBsAg. Alignments of the deduced amino acid sequences obtained from ten clones of per OBI patient. The alignment was performed using one reference HBV sequence genotype H (AY0954.1) with the CLCbio software. The shadow colors indicate the HBsAg domain, as show in the gene S figure on the left side. Residues identical to the three genotype H reference sequences are indicated by dots. Mutations are indicated in the red boxes.
Additional deduced amino acid sequences analysis of the HBsAg. Alignments of the deduced S gene amino acid sequences of the ten clones per OBI patient, with the 98 deduced S-gene amino acid sequences genotype H. Multiple alignments were performed using MEGA 6.06 software. The shadow colors indicate the HBsAg domain, as show in the gene S figure on the left side. Residues identical to the three genotype H reference sequences are indicated by dots. Mutations are indicated in the red boxes.



