



Hepatitis C virus (HCV) has a short replication time, high mutation rates and large population sizes, all of which make it an excellent experimental model for evolution studies, because evolution can be visualized in real-time. In this review, we discuss the implications to study HCV evolution at the interpatient and intrapatient levels of infection. The HCV interpatient dynamics is relatively slow, because the generation time is generally long. Then, at population level, the HCV diversity originated by the high mutation and replication rates is modulated by the bottleneck at transmission. Thus, when the virus is transmitted to other hosts, viral diversity is reduced as a result of the founder effect. On the other hand, during intrapatient infection, HCV evolves rapidly, resulting in quasispecies. Accumulated evidence suggests that this quasispecies composition of the HCV population within the same individual may allow the virus to evade the immune response or escape treatment, leading to chronic infection. Thus, a better understanding of the complexities underlying the molecular evolution of HCV in natural populations is needed before accurate predictions of viral evolution can be made. In summary, HCV evolves both within and among patients. Consequently, HCV evolution should be studied at both levels in order to better understand the natural history of the virus and its potential implications in epidemiology, outcome of infection and progression of liver disease.
Hepatitis C Virus (HCV) is an enveloped single-stranded positive RNA virus1 that currently infects approximately 3% of the world’s population.2 In about 80% of infections, HCV causes a silent chronic hepatic illness that can lead to fibrosis, cirrhosis and hepatocellular carcinoma.3 This RNA virus displays high genetic diversity. Seven genetic clades and various subtypes have been described and are associated either with a specific geographic distribution or with different biological features.4–8 HCV evolves very rapidly during the progress of infection, resulting in genetically diverse viral populations whose composition is determined by a combination of evolutionary processes that include a short generation time, high mutation rates, large population sizes, natural selection and random genetic drift. All these mechanisms allow adaptation of viruses to selective pressures and make HCV an excellent experimental model where evolution can be monitored in real-time.9,10 The evolution of HCV is mostly determined by its own characteristics and by host-mediated or antiviral selection pressures.11–17 Nevertheless, despite their apparent simplicity and advantages, viral populations exhibit features that make their evolutionary dynamics extremely complex to evaluate.11,12 Firstly, in nature, HCV exists in an intricate hierarchy of populations, from single infected cells to global pandemics. Secondly, the long infectious periods, which are in the order of years, mean that both inter- and intrapatient evolutionary patterns can be observed. Additionally, HCV also exists within its hosts as a dynamic pool of genetically distinct but closely related variants referred to as “quasispecies”.11,12 Accordingly, HCV appears to exhibit qualitatively different evolutionary dynamics when its genetic diversity is studied at different levels. In this way, the substitution rate of HCV may differ in intra- and interpatient as well as among the different regions of the genome. Then, its fast adaptation allows this virus to prevail over natural or artificial barriers such as host defenses, pathogeny and drug therapy.13–17
As mentioned above, regardless of their rapid growth, HCV virus populations suffer repeated bottlenecks, both within hosts and when transmitted from host to host. The population bottleneck can be defined as a marked reduction in the number of viral genomes within a population which leads to significant effects. Particularly, intrapatient evolutionary effects of bottlenecks can be observed by comparing sequences of quasispecies that represent different virions from a single chronically infected patient over several years.18,19 On the other hand, in interpatient evolution, the size of the viral population replicating is a very informative point to consider. Additionally, when the virus is transmitted, selection can act deeply in large populations, whereas the random effect of genetic drift is more severe in small populations.
Thus, one of the largest distinctions between these two levels is that interpatient evolution is mostly conducted by numerous bottlenecks arising from transmission events, whereas in intrapatient evolution the bottlenecks are not very relevant while the patient is not treated.20 The two patterns outlined above suggest that different evolutionary and population genetic processes should be inferred from inter- and intrapatient sequence data. For all these reasons, understanding HCV evolution is important itself but also in view of the fact that viral diversity could be relevant in the outcome of the infection and liver disease progression.7,8,21–23
Interpatient EvolutionAs it happened with HIV, HCV may have been transmitted to humans through interspecific infections that were only possible with the demographic changes that occurred 50-100 years ago. The fact that HCV is transmitted mainly through parenteral routes implicates the transmission through unsterilized needles, blood transfusion and injecting drug use (IDU). The current HCV screening and the expansion of needle exchange programs have led to reductions in blood transfusion risk and significant falls in HCV incidence among IDUs.24 For this reason, the HCV interpatient dynamics is relatively slow, since the time between transmission events is generally long (months or years).25 Then, at population level, the HCV diversity originated with high viral mutation and replication rates is modulated by the bottleneck at transmission.26 Each new host represents a novel environment for the virus. Such extreme genetic bottleneck associated with transmission may largely depend on the structure of the virus population in the donor host, the virus genotype, the host factors, and the rapid and radical decline in population size that normally reduces the population’s genetic diversity. Accordingly, bottleneck could be enhanced by the influence of host factors such as the favorable IL28B genotype rs12979860 (CC) that was associated with the spontaneous clearance (SC) of HCV in several studies.27,28 Additionally, the HCV non-1 genotypes have also been related with SC increasing the importance of HCV diversity in the persistence and pathogeny of this virus.29
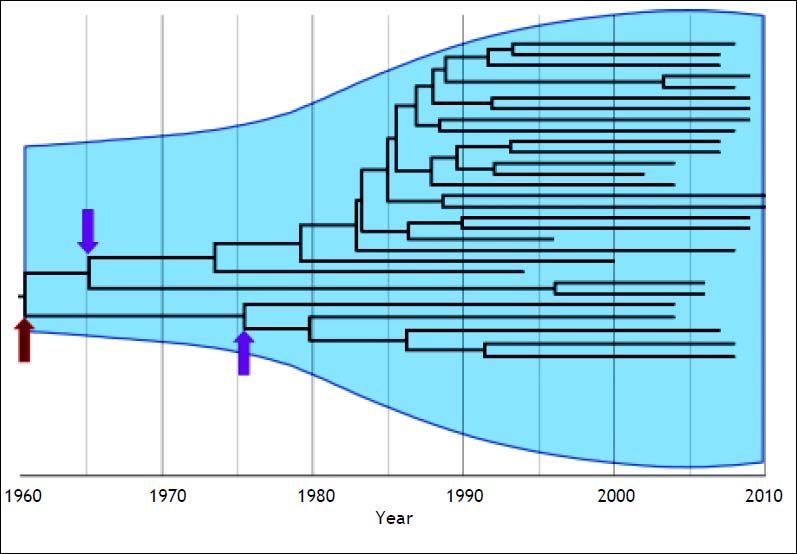
On the other hand, if the structure of virus population is genetically heterogeneous, transmission bottlenecks could be determined partially by the genetic diversity available in the host. Therefore, among-virus competition after transmission typically generates a strong population bottleneck, making it very unlikely that multiple viral lineages will be repeatedly transmitted.30–32 Consequently, the phylogenetic structure of HCV at the population level reflects the demographic and spatial history of transmission.26 In this context, it is known that HCV phylogenetic trees often span several decades so the time when two viral lineages coalesce is close (relative to the timescale of the phylogeny) to the actual transmission times. Accordingly, interpatient phylogenies of HCV sampled through time are not stepwise and show the persistence of multiple lineages through time (Figure 1). The shapes of these trees are primarily determined by neutral demographic and spatial dynamics in both immune-mediated positive selection and purifying selection for replication ability.26–33 In this way, the evolutionary analysis has shed light on the origins of HCV and on the epidemic spread of viral variants in different geographic locations and populations.
. The overall process is depicted by the diversification of the current lineages from the 60’s to the 80’s independently of the actual time of sampling. The arrows indicate possible bottleneck effects. The dark red one indicates the possible introduction of HCV in the community, whereas the magenta ones may indicate the acquisition of the virus by different routes of transmission. Virtually, every transmission event is thought to be a bottleneck effect for virus diversity. The light blue shaded area represents the viral genetic diversity trough the time measured by the Bayesian Skyline methodology.65")
Interpatient evolution of HCV in a country. Maximum Clade Credibility Tree constructed by Bayesian coalescent methodology from direct sequences of the first half of the E2 region in plasma samples from HCV genotype 1a isolations in Argentina. The tree is presented in time scale (calendar years). The overall process is depicted by the diversification of the current lineages from the 60’s to the 80’s independently of the actual time of sampling. The arrows indicate possible bottleneck effects. The dark red one indicates the possible introduction of HCV in the community, whereas the magenta ones may indicate the acquisition of the virus by different routes of transmission. Virtually, every transmission event is thought to be a bottleneck effect for virus diversity. The light blue shaded area represents the viral genetic diversity trough the time measured by the Bayesian Skyline methodology.65
Additionally, HCV diversity has also implications for the transmission. It is known that genotype distribution is associated with the mode of transmission, with subtypes 1a, 3a and 4 being mostly IDU-related and genotypes 1b and 2 associated with blood transfusion and unsafe medical procedures.34–36 Currently, this distribution is changing due to the controls in transfusion procedures, the increment of IDUs, the immigration and the controls in vertical and sexual transmission. A clear example of this is the increasing prevalence of genotype 4 in Europe.36–39 HCV genotype 4 is considered a difficult-to-treat genotype in HCV-monoinfected and HIV/HCV coinfected patients, because the rate of sustained virological response to pegylated interferon plus ribavirin is poorer than that for HCV genotypes 2 or 3.8,40–43 Then, this issue is of importance since the HCV genotype and subtype are also very relevant in terms of antiviral therapy.
Intrapatient EvolutionHCV is among the most successful of all persistent human viruses. Chronic infection is seen in up to 75% of those infected by successfully undermining virus-specific immunity.44 Previous studies suggest that HCV alters the host defense and innate immunity early during infection through a variety of complementary mechanisms, thereby facilitating chronic infection and leaving host defenses to other infectious agents intact.45 The mechanism responsible for the high rate of persistence is attributed to the efficient capacity of generation and fixation of escape mutants associated to HLA in the non structural protein of HCV, which allows the virus to evade the adaptive and innate components of the host’s immune system.46 Additionally, HCV capacity of persistence is strongly influenced by the age and the nature of the host’s innate and adaptive antiviral immunity. In this sense, several studies had shown that IL28B, CXCL10, IFNL3, HLA class I and II are associated with outcome of HCV and may explain an important percentage of spontaneous clearance of HCV infection.47–53 The misincorporation of nucleotides during HCV replication and the subsequent effects of selection pressure, from either neutralizing antibodies or cytotoxic T lymphocytes, as well as the drift that fix substitutions in populations, represent the fundamental driving force in intrapatient evolution.21,54,55 As a consequence, HCV evolves very rapidly during intrapatient infection, resulting in variants whose dynamic composition is determined by a combination of evolutionary processes that include mutation, replication rate, natural selection, and random genetic drift at transmission time. In this sense, accumulated evidence suggests that the genetic variation of HCV within the same individual may allow the virus to evade the immune response, leading to chronic infection.19 Virtually in a patient infected with HCV, it is likely that all possible variants are present in the quasispecies at every moment. However, the point is to determine which the threshold of variant frequency for establishing a particular phenotype is, either a variant resistant to treatment or a variant causing more severe liver damage. Thus, a rapid emergence of resistant variants was detected in the majority of patients with treatment failure to direct-acting antiviral (DAA).56–59 However, this is not an unexpected finding. Naturally, HCV can produce approximately 1012 virus particles per day.60 Furthermore, the viral replication complex lacks proofreading capacity, leading to a large number of mutants. As a result, the virus population is a highly complex and continuously changing mutant spectrum or mutant cloud known as quasispecies.61 These are the lowest level of viral heterogeneity variation with only 1 to 2% and, thus, the major challenge with regard to resistance to treatment.61 Despite the fact that, in theory, an active HCV infection can produce all possible mutants in just one day, not all are able to persist in the population. This is because, in the viral genome, there are constraints with restrictions to change since most mutations generate viral variants that have a reduced replicative ability and, therefore, do not proliferate. However, this will surely be different for each patient because of the multiplicity of factors involved in each process related to viral infection.
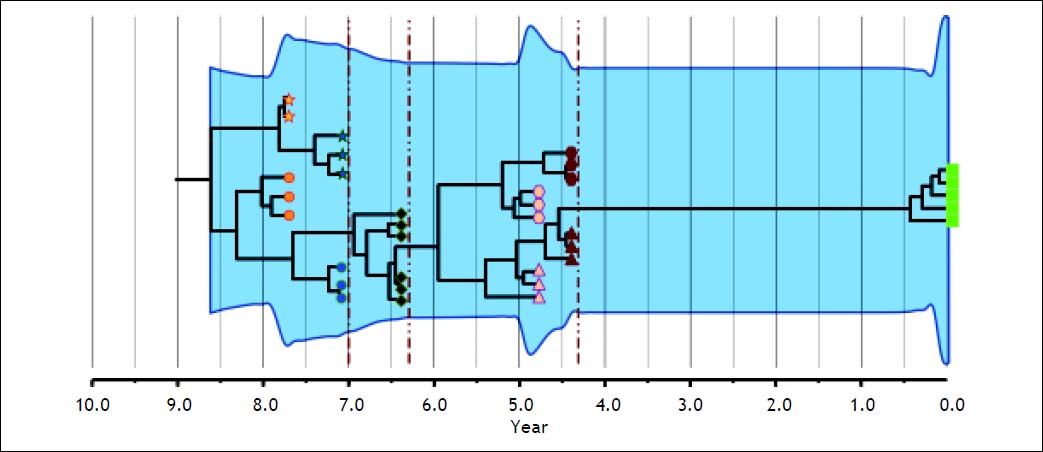
Phylogenetic analyses of intrapatient evolution reconstructed from HCV genomes sampled from multiple time points have mostly revealed two or more genetically distinct co-existing viral lineages that are not detected at all sampling times (Figure 2). This phenomenon is seen in both acute62 and chronic infection.17,63–65 Then, the intrapatient tree topology is manifested by a temporal structure that shows continual adaptation to the patient immune responses and by high rates of lineage extinction which result in a modest genetic diversity at any time point. This suggests that the succeeding viral population is not always immediately derived from the preceding population and sometimes would be the effect of evolutionary shift. Briefly, regardless of the complexity or diversity of the viral populations at the early stage of infection, HCV evolves towards genetic diversification following selective pressure. Once the environment becomes stable, the entire population tends towards homogeneity.18 Then, HCV intrapatient evolution, which may be driven by adaptive selection, becomes important from a clinical perspective because it contributes to the variability in disease progression and development of drug resistance. In this sense, pathobiological studies linking nucleotide and amino acid sequences with clinical findings have identified relationships between certain genotypes and characteristic biological properties.23 For example, HCV-3 infection was found to be associated with a high level of liver steatosis.66 At quasispecies level it has been reported that an increase in HVR1 diversity during acute infection predicts progression to chronic disease, whereas a decrease correlates with resolution of the disease.21
. Each symbol (star, circle, diamond, hexagon, triangle and square) represents different viral lineages. The colors indicate, as the axis position does, the time of the sampling. The vertical dark red dotted lines represent bottleneck events where one of the lineages (shapes) got extinct. The overall process is depicted by the modification or extinction of existing lineages (quasispecies shift). The light blue shaded area represents the viral genetic diversity trough the time measured by the Bayesian Skyline methodology.65")
Intrapatient evolution of HCV in an 8-year follow up. Maximum Clade Credibility Tree constructed by Bayesian coalescent methodology from cloned sequences of the first half of the E2 region in serially obtained plasma samples from a single patient. The tree is presented in time scale (years before present). Each symbol (star, circle, diamond, hexagon, triangle and square) represents different viral lineages. The colors indicate, as the axis position does, the time of the sampling. The vertical dark red dotted lines represent bottleneck events where one of the lineages (shapes) got extinct. The overall process is depicted by the modification or extinction of existing lineages (quasispecies shift). The light blue shaded area represents the viral genetic diversity trough the time measured by the Bayesian Skyline methodology.65
On the other hand, it has been extensively described the effect of genetic variability between HCV genotypes and subtypes on response to therapy. HCV-1 and 4 were found to be more resistant to interferon based therapies than HCV-2 and 3 and, in the same way, HCV-1b respond better than HCV-1a.67,68 However, there is contradictory data about the influence of quasispecies on treatment response.69–72 Some reports have demonstrated that resistant mutants are present in DAA naïve-patients as either dominant or minority species.56,73 When treated with DAA, resistant variants, initially present as minority species, may expand to occupy the free replicative space, thus becoming the dominant variant.72,74 Consequently, this may lead to failure of antiviral regimen. In this context, it has been reported that mutations need to be present at a certain frequency to result in a measurable phenotypic effect.75,76 However, more recently, literature has emerged that offers contradictory findings about this topic.72,77 For all these reasons, the study of viral genetic variation and the emergence of drug resistance are of major importance in the era of new treatments.
ConclusionIn intrapatient evolution, once the virus is installed in the new host, the first molecular events occurring immediately after may result in the generation of a highly heterogeneous viral population.20,26 Subsequently, the HCV specific adaptive immunity probably shapes this founder population into a dynamic quasispecies complex, each of which may become dominant at different phases of HCV infection. Then, negative selection acts to achieve a stable adaptation to the host. HCV evolution is therefore defined mainly by two distinct mechanisms: drift and shift on the variants present in a chronic infected patient. This consideration implies that succeeding dominant subpopulations may not be directly derived from each other but may rather share a common ancestor.
Lastly, in the interpatient evolution, the transmission to a new host represents a novel environment for the virus. Such extreme genetic bottleneck may largely depend on the inoculum and the structure of the virus population in the donor host.
In conclusion, the long infectious period of HCV means that evolution occurs both within and among patients. Consequently, the evolution of HCV should be studied both at interpatient level (bottleneck at transmission) to better understand the natural history of the virus and its potential implications in epidemiology and, at intrapatient level (adaptation to the exogenous and host’s pressures) to better understand the outcome of infection and the progression of liver disease.
SupportThis work was supported by the Agencia Nacional de Investigaciones Científica y Técnicas PICT 0705, Universidad de Buenos Aires UBACyT 405, Consejo Nacional de Investigaciones Científicas y Técnicas CONICET PIP 0215.
AcknowledgmentWe thank Victoria Eusevi and Paula S. Perez for enhancing the readability of this paper.



