








Background. Intestinal mucosal barrier dysfunction in liver cirrhosis and its implicated mechanisms is of great clinical importance because it is associated with the development of serious complications from diverse organs through promotion of systemic endotoxemia.
Aim. The present study was designed to investigate whether enterocytes’ proliferation, apoptosis and intestinal oxidative stress are altered in the intestinal mucosa of patients with compensated and decompensated liver cirrhosis.
Material and methods. Twelve healthy controls (group A) and twenty four cirrhotic patients at a compensated (n = 12, group B) or decompensated condition (n = 12, group C) were subjected to duodenal biopsy. In intestinal specimens mucosal apoptotic and mitotic activity and their ratio were recorded by means of morphological assessment and mucosal lipid hydroperoxides were measured. Plasma endotoxin concentration, an index of gut barrier function, was also determined.
Results. Cirrhotic patients presented significantly higher serum endotoxin concentrations as compared to healthy controls (P < 0.001), whilst endotoxemia was higher in decompensated disease (P < 0.05 vs. compensated cirrhosis). Intestinal mucosal mitotic count was significantly lower in patients with compensated and decompensated cirrhosis compared to controls (P < 0.01, respectively), whilst a trend towards increased apoptosis was recorded. The mitotic/apoptotic ratio was significantly reduced in groups B (P < 0.05) and C (P < 0.01) as compared to controls. Intestinal lipid peroxidation was significantly increased in decompensated cirrhotics (P < 0.001 vs. groups A and B).
Conclusions. The present study demonstrates for the first time that human liver cirrhosis is associated with decreased intestinal mucosal proliferation and proliferation/apoptosis ratio even at early stages of cirrhosis and increased intestinal oxidative stress in advanced liver disease.
The liver is continuously exposed to gut microbiome-associated and immunologically active molecules, such as lipopolysaccharide, via the portal vein supply. This lifelong immunological cross-talk in health and disease is described by the term gut-liver axis, which emphasizes the role of intestinal structural and functional alterations in the pathogenesis of liver diseases.1 Experimental and clinical studies have demonstrated that liver cirrhosis is associated with intestinal barrier dysfunction and increased gut permeability.2–4 Intestinal barrier dysfunction in liver cirrhosis and its implicated mechanisms is of great clinical importance because it is associated with the development of serious complications from diverse organs through promotion of systemic endotoxemia.5–9
The intestinal epithelium consisted of a monolayer of epithelial cells linked close to the apical surface by the tight junctions seem to be the most important factor in determining intestinal permeability.10 We have recently shown for the first time in humans that liver cirrhosis induces significant alterations of enterocytes’ tight junctions, which is an important pathogenetic mechanism of intestinal hyperpermeability.11 On the other hand, mucosal barrier is also dependent on epithelial homeostasis that is determined by the balance between cell proliferation and death, which regulate intestinal cell turnover and mucosal cellularity.
Oxidative stress is not only a causative factor of cellular injury but also a pivotal regulator of all crucial cellular processes including metabolism, growth, differentiation and death.12 Directly or indirectly it is implicated in all major physiological and pathological processes and plays a pathogenic role in diverse types of intestinal injury.13–16 Experimental studies have demonstrated that oxidative stress contributes to the structural and functional derangement of the intestinal mucosal barrier in liver cirrhosis.17,18
The present study was designed to investigate whether enterocytes’ proliferation and apoptosis and intestinal oxidative stress are altered in the intestinal mucosa of patients with compensated and decompensated liver cirrhosis.
Material and MethodsPatientsThe study was approved by the Ethics Committee of Patras University Hospital, Patras, Greece. Written informed consent was obtained from all subjects enrolled in the study.
Patients were eligible for inclusion in this prospective study if they had been diagnosed with liver cirrhosis (viral, alcoholic or other aetiology). The diagnosis of liver cirrhosis was established by means of histology and/or by its clinical, laboratory, endoscopic, or imaging manifestations. Cirrhosis severity was assessed according to the Child-Pugh classification.
The exclusion criteria were: malignancy, diabetes mellitus, rheumatic diseases, renal diseases, gastrointestinal diseases (e.g. celiac disease, inflammatory bowel disease, gastrointestinal bleeding the last four weeks) or intestinal surgery, infections included spontaneous bacterial peritonitis (in the last four weeks), alcohol abuse in the last four weeks, elevated serum or urine amylase and treatment during the last month with medications that might affect intestinal oxidative stress like antibiotics, corticosteroids, non-steroid anti-inflammatory drugs and antioxidants (vitamins C and E, allopurinol, N-acetyl-cysteine).
The control group comprised of healthy individuals matched for age and sex, without any of the above stated exclusion criteria, who underwent an upper gastrointestinal tract endoscopy without pathologic findings (group A, n = 12). Cirrhotic patients were subsequently divided into two groups: patients with compensated cirrhosis (group B, n = 12,) or decompensated cirrhosis (group C, n = 12). Classification in decompensated or compensated disease was based on Child-Pugh class; Child-Pugh A was classified as compensated cirrhosis whereas Child-Pugh B or C as decompensated cirrhosis.
All subjects enrolled in the study underwent an upper gastrointestinal tract endoscopy, during which biopsies were obtained from the second portion of the duodenum, distal to the ampulla of Vater.
Endotoxin measurementsBefore endoscopy, all subjects enrolled in the study were subjected to blood sampling from a peripheral vein for endotoxin measurement. Blood samples were collected in endotoxin-free vials and then plasma was separated and stored at −80°C until processed. Endotoxin concentration was determined by the quantitative chromogenic Limulus Amebocyte Lysate test (LAL, QCL-1000, Lonza, Walkersville, MD, USA) and expressed in EU/mL. Samples were processed according to the manufacturer’s instructions.
Intestinal lipid hydroperoxides (LOOH) assayIntestinal samples were homogenized in 0.1 mL 10 mM phosphate buffer, pH 7.2 (using a microtube fitting pestle), saving 10 µL for protein quantification.19 Homogenate LOOH were extracted with 0.09 mL chloroform:methanol (2:1 v:v) followed by centrifugation (at 15,000 g for 5 min), chloroform layer vacuum-drying, and LOOH solubilization with 0.25 mL methanol. LOOHs were estimated with the FOX assay.20 Measurements were performed by two researchers (DZ, CDG) who were unaware of the clinical data.
Histopathological evaluation of proliferation and apoptosis in the intestinal mucosaDuodenal biopsies were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 4 µm and stained with hematoxylin and eosin. In each intestinal mucosal specimen, mitoses (Figures 1A–1B) and apoptotic bodies (Figure 1C) of the cryptal epithelium were identified and tallied using a morphometric analysis, which has been described in detail elsewhere.21 Apoptotic bodies were defined as rounded vacuoles with fragments of karyorrhectic nuclear debris and were differentiated from small isolated fragments of nuclear chromatin and intraepithelial neutrophils. Apoptotic bodies and mitoses were counted in all architecturally successive crypts included in the specimen, regardless of crypt orientation, and their total number was divided by the number of the crypts. The number of apoptotic bodies per crypt is referred to as the apoptotic body count and the number of mitoses per crypt as the mitotic count. Histological analyses were performed by two experienced pathologists (ACT, CDS) who were unaware of the clinical data.
 in the intestinal epithelium of a healthy subject (A) and in a patient with decompensated cirrhosis (B). Detection of apoptotic bodies (red arrows, H&E x 200) in the intestinal epithelium of a patient with decompensated cirrhosis (C).")
Microphotographs showing mitoses (red arrows, H&E ×200) in the intestinal epithelium of a healthy subject (A) and in a patient with decompensated cirrhosis (B). Detection of apoptotic bodies (red arrows, H&E x 200) in the intestinal epithelium of a patient with decompensated cirrhosis (C).
Data were analyzed using the SPSS statistical package (SPSS Inc, 2001, Release 11.0.0, USA). Normality of data was tested using the Shapiro-Wilk Test. Comparisons were performed using the nonparametric analysis of variance (Kruskal-Wallis test) followed by a post-hoc Mann-Whitney U test. In all cases, a P-value < 0.05 was considered as significant.
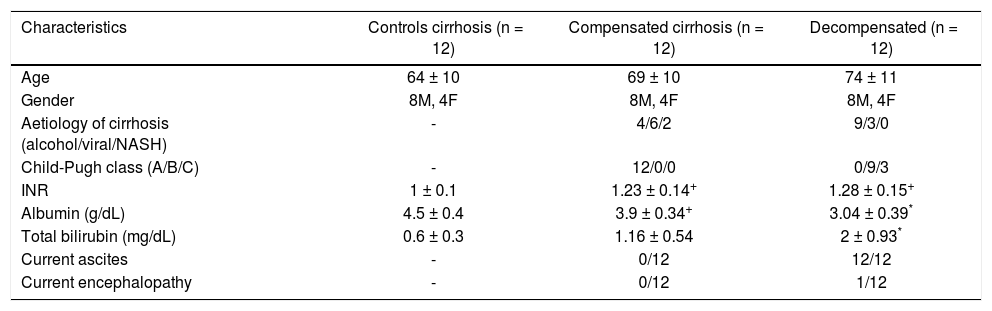
ResultsPatients’ characteristicsCharacteristics of the enrolled population are summarized in table 1. No significant differences with regard to age and gender were observed among groups. The causes of liver cirrhosis were alcohol in 13 patients, viral infection in 9 patients and non-alcoholic steatohepatitis in 2. In all, 12 patients were classified as Child-Pugh class A, 9 as Child B, and 3 as Child C. All patients with decompensated cirrhosis had ascites whereas ascites was absent in all patients with compensated disease.
Characteristics of patients with compensated or decompensated liver cirrhosis and healthy controls.
| Characteristics | Controls cirrhosis (n = 12) | Compensated cirrhosis (n = 12) | Decompensated (n = 12) |
|---|---|---|---|
| Age | 64 ± 10 | 69 ± 10 | 74 ± 11 |
| Gender | 8M, 4F | 8M, 4F | 8M, 4F |
| Aetiology of cirrhosis (alcohol/viral/NASH) | - | 4/6/2 | 9/3/0 |
| Child-Pugh class (A/B/C) | - | 12/0/0 | 0/9/3 |
| INR | 1 ± 0.1 | 1.23 ± 0.14+ | 1.28 ± 0.15+ |
| Albumin (g/dL) | 4.5 ± 0.4 | 3.9 ± 0.34+ | 3.04 ± 0.39* |
| Total bilirubin (mg/dL) | 0.6 ± 0.3 | 1.16 ± 0.54 | 2 ± 0.93* |
| Current ascites | - | 0/12 | 12/12 |
| Current encephalopathy | - | 0/12 | 1/12 |
M: males. F: females. NASH: non alcoholic steatohepatitis.
Patients with decompensated and compensated cirrhosis presented significantly higher endotoxin values in peripheral blood as compared to healthy controls (P < 0.001, respectively). Patients with decompensated disease had significantly higher plasma endotoxin concentrations as compared to compensated cirrhotics (P < 0.05) (Figure 2).
Intestinal lipid hydroperoxides. *P < 0.001 vs. controls, +P < 0.05 vs. compensated cirrhosis.")
Intestinal lipid peroxidation was significantly increased in decompensated cirrhotics (P < 0.001 vs. groups A and B), as shown in figure 3.
Intestinal mucosal mitotic and apoptotic activity. *P < 0.00 vs. control and compensated cirrhosis.")
The values of intestinal mucosal mitotic count, apoptotic body count and their ratio are shown in table 2. Mucosal mitotic count was significantly lower in patients with compensated and decompensated cirrhosis compared to controls (P < 0.01, respectively), whilst a trend towards increased apoptosis was recorded. The mitotic/apoptotic ratio was significantly reduced in groups B (P < 0.05) and C (P < 0.01) as compared to controls. These alterations are graphically presented in figure 4.
Intestinal mucosal mitotic count, apoptotic count and their ratio.
| Group A (n = 12) | Group B (n = 12) | Group C (n = 12) | P value | |
|---|---|---|---|---|
| Mitotic count | 0.135 (0.047–0.161) | 0.062 (0.032–0.095) | 0.049 (0.025–0.214) | I/II: 0.009, I/III: 0.007 |
| Apoptotic count | 0.027 (0.010–0.062) | 0.028 (0.015–0.120) | 0.038 (0.021–0.071) | NS |
| Mitotic/apoptotic ratio | 3.677 (1.342-15) | 2 (0.666-6.333) | 1.875 (0.074-3.333) | I/II: 0.036, I/III: 0.005 |
Values are medians (min-max). Group A: control, Group B: compensated cirrhosis, Group C: decompensated cirrhosis.

Increased intestinal permeability has been repeatedly confirmed in experimental cirrhosis and the clinical setting as well and is considered as the starting and continuously promoting event for the development of spontaneous bacterial peritonitis and other complications of cirrhosis, even from distant organs, through systemic endotoxemia.2–4,22 The intestinal epithelium seems to be the most important factor in determining intestinal permeability and we have recently demonstrated for the first time that the expression of the TJ-associated proteins occludin and claudin-1 is decreased in the intestinal epithelium of patients with liver cirrhosis contributing, at the cellular level, to increased paracellular permeability and gut barrier dysfunction.11
Other factors of indisputable importance in the determination of gut barrier function integrity are intestinal epithelial cell proliferation and apoptosis, which are normally in a continuous homeostatic process. The present study showed a significant reduction in intestinal epithelial cell proliferation and a trend towards increased apoptosis, which did not reach statistical significant levels. However, the proliferation/apoptosis ratio in the intestine of patients with liver cirrhosis was found significantly decreased. These cellular alterations demonstrate an important imbalance of intestinal homeostasis, which might compromise the integrity of the mucosal barrier through decreased mucosal cellularity and defective healing response to diverse injurious insults.
Mucosal cellular alterations were simultaneously detected in patients with compensated and decompensated liver cirrhosis, whilst no significant differences existed between these groups. This finding might explain the existence of gut barrier dysfunction and significant endotoxemia in both groups of cirrhotic patients. Previous studies have also shown that gut barrier dysfunction and intestinal hyperpermeability occurs early in the course of liver disease.23 The greater magnitude of gut-derived endotoxemia observed in patients with decompensated disease could be attributed to additional factors contributing to gut barrier derangement in this group, such as the greater reduction of TJs proteins expression, as shown in our previous study,11 and the existence of high oxidative stress detected in the present study.
Previous studies have shown a close association of liver cirrhosis with oxidative stress. In humans, oxidative stress has been detected in the blood and liver tissue of cirrhotic patients.24,25 Oxidative stress-mediated hepatic mitochondrial dysfunction has been previously shown in cirrhosis and assessment of this parameter via 13C-based breath tests seems to be a promising non-invasive technique for evaluation of liver function.26 Regarding the intestinal mucosa in liver cirrhosis, high levels of oxidative stress have been previously shown in experimental animals.17,18 To the best our knowledge, this is the first study demonstrating the presence of high oxidative stress in the intestinal mucosa of patients with decompensated cirrhosis. In the present study high intestinal mucosa oxidative stress was detected only in patients with decompensated cirrhosis, which is in line with previous reports demonstrating high blood oxidative stress in patients with advanced cirrhosis.27 A potential explanation might be the higher levels of systemic endotoxemia observed in decompensated disease, which is an important oxidative stress promoter through activation of a systemic inflammatory response, and the depleted hepatocyte antioxidant defenses in advanced liver disease. Potential causes of high intestinal oxidative stress in decompensated cirrhosis are enterocyte mitochondria and increased activity of mucosal xanthine oxidase in conjunction with decreased activity of antioxidant enzymes like xanthine dehydrogenase, superoxide dismutase and catalase.17,28
In the present study, intestinal cellular alterations in compensated cirrhotics occurred without concurrent high intestinal oxidative stress, which indicates that other factors, such as systemic endotoxemia, might be implicated in intestinal mucosal abnormalities at early stages of liver disease. However, in decompensated cirrhosis high intestinal oxidative stress could have exerted a pivotal role in the further impairment of mucosal barrier integrity through inhibition of cell proliferation,29 as shown in the present study, and through disruption of the tight junction structural complex,30 as we have previously shown. A difficult to explain finding of the present study was that oxidative stress, a wellknown promoter of apoptosis, did not induced a significant increase of intestinal apoptosis in decompensated liver disease (P = 0,07 vs. group A). A potential explanation might be the potential lower sensitivity of the solely morphological assessment of apoptosis used in the present study.14
In conclusion, the present study demonstrates for the first time that liver cirrhosis in humans is associated with significant alterations of intestinal epithelial homeostasis, as evidenced by reduction of mucosal proliferation and proliferation/apoptosis ratio. Decompensated disease was associated with high levels of intestinal oxidative stress, potentially contributing to intestinal injury and higher degree of systemic endotoxemia in these patients. These changes might represent important mechanisms, at the cellular level, for intestinal barrier dysfunction and hyperpermeability in patients with liver cirrhosis. Therapeutic agents that restore intestinal epithelial homeostasis and reduce intestinal oxidative stress, like enteral immunonutrition (glutamine), probiotics, antioxidants and gut regulatory peptides (bombesin, neurotensin) might be of potential value in patients with liver cirrhosis.31–33,14



