



Background. Liver involvement in celiac disease (CD) varies from asymptomatic mild non-specific hepatitis to liver failure. Here we report the first child with liver failure due to a sclerosing cholangitis associated with CD.
Case report. An 11 year old girl presented with fatigability for 1 year and jaundice and abdominal distension for 3 weeks. On examination, the growth parameters were below 3rd percentile; she had splenomegaly and severe ascites. Liver function tests revealed elevated liver enzymes (ALT 84 U/L, total bilirubin 98.7 µmol/L, and direct 58.3 µmol/L, gamma-glutamyltransferase 111 U/L, INR 2.7, and albumin 16 g/L). Extensive investigations excluded infectious, metabolic, structural, and endocrine causes of chronic liver disease. Because of the short stature and anemia, CD was suspected, and serological evaluation revealed increased IgA antibodies to tissue transglutaminase (385 units; normal, 0–20 units). Histopathological examination of small intestinal biopsies showed total villous atrophy consistent with celiac disease. Liver biopsy showed bridging fibrosis, portal tract expansion by lymphocytes, plasma cells, and neutrophils, bile ductular proliferation, and periductular fibrosis. Magnetic resonance cholangiography revealed beading and narrowing appearance of intra- and extrahepatic bile ducts. The histopathological and imaging findings are diagnostic of sclerosing cholangitis. The child was initiated on ursodeoxycholic acid, gluten free diet for life, and steroid that was tapered over 3 months. At 3 month follow up, liver function tests completely normalized.
Conclusion. CD is a potentially treatable cause of liver failure. All patients with severe unexplained liver disease should undergo serological screening for CD.
Celiac disease (CD) is an immune mediated enteropathy caused by permanent gluten intolerance.1 The liver abnormality in CD has a wide spectrum ranging from mild hepatic abnormalities to severe liver disease.2–8 The liver disease may be a component of the CD and present as a cryptogenic acute or chronic hepatitis with or without cholestasis. This cryptogenic form is usually characterized by predominant serum aminotransferase abnormalities, and it has been reported in about 40% of adults and 54% of children with CD at the time of diagnosis.6,7 It is usually reversible on a gluten free diet (GFD).3,9,10
Alternately, the CD may be concurrent with the autoimmune liver disease, but not the cause of it.5 The CD in this autoimmune form is commonly asymptomatic or over-shadowed by the manifestations of the autoimmune liver disease. Its contribution to the severity of the autoimmune liver disease is uncertain, and the impact of GFD on the liver damage is controversial.5,11
There are only a few papers in the literature on the occurrence of liver failure in untreated celiac patients.2,9,10,12 The underlying liver pathology in most cases has been non-specific, and the role of the GFD in such circumstances has been unclear. Here we report a child with liver failure due to sclerosing cholangitis associated with CD. Liver function tests normalized on a combined treatment using prednisone, ursodeoxycholic acid, and GFD, and they have remained normal for 4 years on GFD and ursodeoxycholic acid.
Case ReportAn eleven-year old girl was referred to our service in August 2007 for further evaluation for decompensated liver disease. She had developed increasing abdominal distension and jaundice one month prior to admission associated with mild itching. Anorexia, fatigability, and poor growth had been present for 3 years. There was no history of diarrhea, vomiting, fever, herbal or drug ingestion.
On physical examination, both weight and height were below 3rd percentile. She looked pale, wasted, and icteric with clubbed fingers and palmar erythema. Abdominal examination revealed gross ascites, splenomegaly, and an impalpable liver.
Laboratory tests showed the following: hemoglobin 9.4 gm/L, white cell count 5.7 x 10, platelets count 156 x 103, erythrocyte sedimentation rate 110 mm/h, total serum bilirubin 98.7 (normal, 3-17 µmol/L), direct bilirubin 58.3 (normal, 0-3 µmol/L), alanine aminotransferase 200 (normal, 30-65 U/L), alkaline phosphatase 458 IU/L, gamma-glutamyl-transferase 111 (normal, 7-32 U/L), albumin 16 g/l (35-50 gm/L) and INR 2.7.
Extensive laboratory evaluations excluded Wilson disease, viral hepatitis, metabolic diseases, endocrine diseases, and alpha -1-antitrypsin deficiency. Antinuclear antibodies, smooth muscle antibodies, anti-liver/kidney microsomal antibodies, anti-mitochondrial antibodies, and perinuclear antineutrophilic cytoplasmic antibodies were negative. Serum immunoglobulin levels were elevated: immunoglobulin G 23 gm/L (normal 3.7-15 gm/L).
Abdominal ultrasonography showed a markedly heterogeneous, coarse, shrunken, and nodular liver, mild splenomegaly, and gross ascites. Vitamin K, 5 mg intravenously, failed to correct the coagulopathy. Fresh frozen plasma partially corrected the coagulopathy to INR 1.8. Two liters of ascitic fluid were removed, and analysis revealed a transudate that was negative by gram stain, culture, and cytology.
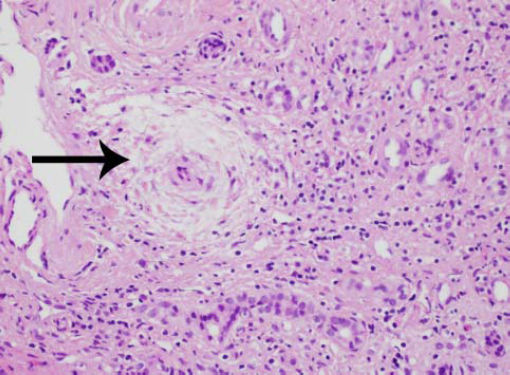
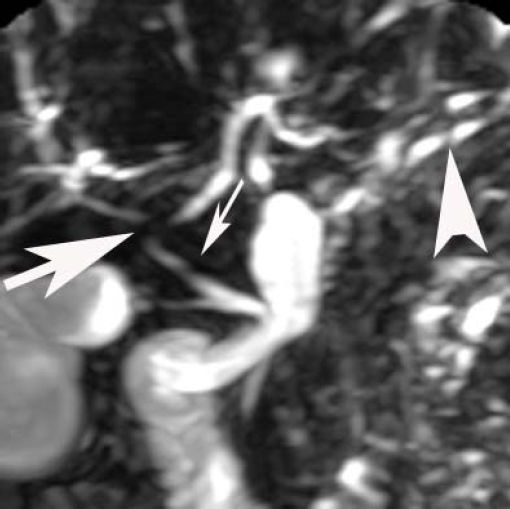
Laparascopically obtained liver biopsy tissue showed bridging fibrosis, nodule formation, portal tract expansion by lymphocytes, plasma cells, and neutrophils, bile ductular proliferation, and periductular fibrosis and edema (Figure 1). Magnetic resonance cholangio-pancreatography revealed beading and narrowing of the intra and extra hepatic bile ducts consistent with sclerosing cholangitis (Figure 2). Because of the short stature and anemia, CD was suspected, and serological evaluation revealed increased IgA antibodies to tissue transglutaminase (385 units; normal, 0-20 units). Histopathological examination of small intestinal biopsies showed total villous atrophy and crypt hyperplasia. The findings were consistent with celiac disease
 (Hematoxylin and eosin, X 100).")
, intrahepatic bifurcation of common hepatic duct (large arrow) and intrahepatic bile ducts (arrow head).")
Uncertainty about the causative relationship between the liver disease and the celiac disease led to combined treatment with prednisone (1 mg/kg/day), ursodeoxycholic acid (20 mg/kg/day), and GFD. The prednisone dose was tapered over 3 months to 2.5 mg daily and discontinued at 6 months. The patient remained on ursodeoxycholic acid and GFD. At 3 months, liver enzymes and synthetic function tests had normalized completely. The celiac profile was fully negative at 6-months.
After four years, liver function tests had remained normal, including serum bile acid and gamma-glutamyltransferase levels; the celiac profile had continued to be negative, and repeated ultrasound examinations of the liver continued to show a heterogeneous, coarse, shrunken, and nodular liver. Serial follow up screening for liver autoantibodies had also remained negative.
DiscussionThe liver injury in CD has a wide spectrum ranging from mild hepatic abnormalities to severe liver disease,2–8 and two clinical forms (cryptogenic and autoimmune liver disease) have been associated with CD.5–7 The response of the liver disease to GFD depends in part on whether the CD accompanies or causes the liver dysfunction. CD that is a concomitant immune disorder in patients with autoimmune liver disease has not been greatly influenced by GFD,5,12 and it has required drug therapies (immunosuppressive agents, ursodeoxycholic acid, or both) that are appropriate for the particular liver disease. In contrast, the cryptogenic form, which is considered to be strictly associated with gluten enteropathy, can present as a mild or severe liver disease in both children and adults.6–11 The histological findings of nonspecific reactive hepatitis (“celiac hepatitis”) that typify this form of liver disease usually resolve after 6–12 months of GFD.6,7 Rarely, the cryptogenic form presents as severe liver disease characterized by cirrhosis11 or liver failure,2,9–11 but even in this advanced stage adherence to a GFD can avoid liver transplantation.2,9 CD could be diagnosed at the presentation or later in the course of liver disease. On occasions, non-compliance on GFD has led to severe liver damage between 1 and 24 months after the diagnosis of CD.9 Re-institution of GFD led to the reversal of the severe liver damage, without the necessity for any medical therapy or liver transplantation.9
Our case had advanced severe liver disease, cholangiographic abnormalities suggestive of sclerosing cholangitis, and histological findings of bridging fibrosis and nodule formation. The clinical phenotype of our patient (female child, no inflammatory bowel disease, cholestatic histological features, negative liver autoantibodies, and abnormal findings on magnetic resonance cholangiography) does not allow an easy categorization. The principal diagnostic considerations included primary sclerosing cholangitis (PSC), autoantibody-negative autoimmune hepatitis (AIH), overlap syndrome of PSC and AIH, and CD alone or in concert with PSC or AIH. The diagnostic uncertainty and the severity of the liver disease justified a treatment regimen that addressed all diagnostic considerations, and prednisone, ursodeoxycholic acid and GFD were instituted. The prompt and complete response to this combination regimen within 3 months and the ability to fully discontinue prednisone after 3 months and still maintain normal tests and well-being 4 years later strongly suggested that CD was the principal basis for the liver disease.
Primary sclerosing cholangitis can affect children in the absence of inflammatory bowel disease, and liver tests can improve during therapy with ursodeoxycholic acid.13 The prolongation of transplant-free survival by such therapy has not been proven in children;14 PSC is typically progressive in children;14,15 and sustained full resolution of the disease, as is our patient, is unusual.15,16 Disappointing results with immunosuppressive therapy have also been reported in children with sclerosing cholangitis.17 Furthermore, perinuclear antineutrophilic cytoplasmic antibodies are commonly present in patients with PSC or AIH,18 and they were not detected in our patient.
Autoimmune sclerosing cholangitis has been recognized in children with clinical phenotypes of classical AIH in the absence of inflammatory bowel disease, and these patients can respond fully to corticosteroid treatment.19,20 Unlike our patient, they usually have the serological markers of AIH, including antinuclear antibodies, smooth muscle antibodies, or anti-liver/kidney microsomal antibodies, and they infrequently remain asymptomatic with normal liver tests after corticosteroid withdrawal.19 Similarly, AIH may occur in the absence of the conventional serological markers and respond fully to corticosteroid treatment, but relapse after corticosteroid withdrawal typifies the diagnosis.21 The completeness of the response and its continued resolution on maintenance GFD after corticosteroid withdrawal in our patient argue strongly for CD as a cause rather than an accompaniment of the liver disease, and it justifies consideration of including “celiac-associated sclerosing cholangitis” within the evolving spectrum of CD-related liver disease. Confirmation of the diagnosis would require discontinuation of the GFD and demonstration of recurrent CD and liver disease. This is an obviously unacceptable course of action.
The advanced liver disease in our patient is best explained by CD, and she exemplifies the cryptogenic form of the chronic liver disease associated with this condition. Five other pediatric cases of celiac disease-related liver failure have been reported in the literature.8,9 Autoimmune hepatitis type 1 was the underlying cause of liver failure in one case,9 whereas the underlying liver pathology was cryptogenic in the remaining four cases.8 The present case represents the first report of liver failure during childhood that strongly implicates a sclerosing cholangitis associated with CD.
The designation of “celiac-associated sclerosing cholangitis” has not been applied previously, but the term “celiac-associated autoimmune cholangitis” has been proposed in adults.22–24 Furthermore, we cannot exclude the possibility of sclerosing cholangitis in these early reports involving three adults with CD and cholestatic hepatitis in the absence of liver failure.22–24 Two of these adults responded to immunosuppressive therapy and GFD23, 24 and the third improved on strict GFD.22
The importance of corticosteroids and ursodeoxycholic acid in the management of “celiac-associated sclerosing cholangitis” is unclear, and the empiric use of these medications in our case was justified by the severity of the liver disease, the associated cholangiographic findings suggestive of sclerosing cholangitis, and the uncertainty that GFD alone could reverse the manifestations of progressive liver failure in a rapid and complete fashion.
ConclusionCD is a potentially treatable cause of liver failure, and its presentation can include cholangiographic abnormalities suggestive of sclerosing cholangitis. All patients with severe unexplained liver disease should undergo serological screening for CD.
Abbreviations- •
AIH: Autoimmune hepatitis.
- •
CD: Celiac disease.
- •
GFD: Gluten free diet.
- •
PSC: Primary sclerosing cholangitis.
Authors disclose no conflict of interest.
FundingAuthors have no financial relationships to disclose.



