



Hepatocellular carcinoma (HCC) is one of the most malignant digestive tumors, and its insidious onset and rapid progression are the main reasons for the difficulty in effective treatment. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) is a key enzyme that regulates phospholipid metabolism of the cell membrane. However, the mechanism by which LPCAT1 regulates HCC metastasis remains unknown. This study aimed to explore its biological function and potential mechanisms concerning migration and invasion in HCC.
Materials and methodsLPCAT1 expression in HCC tissues and its association with clinical outcomes were investigated by western blotting and bioinformatic methods, respectively. The role of LPCAT1 in migration and invasion was assessed via Transwell assays. The expression pattern of epithelial-mesenchymal transition (EMT) markers was quantified by western blotting. The biological behaviors of LPCAT1 in vivo were evaluated using xenograft tumor models and caudal vein metastatic models. Signaling pathways related to LPCAT1 were predicted using gene set enrichment analysis (GSEA) and further confirmed by western blotting.
ResultsLPCAT1 expression was significantly upregulated in HCC tissues and indicated a poor prognosis of HCC patients. Several EMT-related markers were found to be regulated by LPCAT1. HCC cells overexpressing LPCAT1 exhibited remarkably high migration and invasion capacities, upregulated expression of mesenchymal markers and reduced E-cadherin expression. In vivo, LPCAT1 promoted HCC pulmonary metastasis. Furthermore, the Wnt/β-catenin signaling pathway was confirmed to be activated by LPCAT1.
ConclusionsLPCAT1 could serve as a promising biomarker of HCC and as a novel therapeutic target for the treatment of metastatic HCC.
According to the World Health Organization assessment, liver cancer will be responsible for more than 1 million cancer-related deaths worldwide by 2030 [1]. Accounting for the majority of primary liver cancers, hepatocellular carcinoma (HCC) is one of the most frequently diagnosed tumors of the digestive system and has the second-highest case-fatality rate among malignant tumors throughout the world [2]. However, there are currently no specific drugs that can successfully treat HCC, especially for metastatic HCC. Thus, it is essential to urgently elucidate the mechanism of metastasis in a systematic manner, considering that the five-year overall survival (OS) rate of HCC is still less than 70% despite the availability of advanced treatments [3].
As one of the most important hallmarks of cancer, epithelial-mesenchymal transition (EMT) contributes to the rapid progression and poor prognosis of HCC. EMT is a biological cell program that shifts cells from an epithelial phenotype to a mesenchymal phenotype and has been widely studied in recent years. Studies have confirmed that EMT plays a key role in neoplastic metastasis from initiation to completion, which is closely related to poor prognosis [4]. In addition to its role in metastasis, abnormal activation of EMT is a significant mechanism involved in tumor formation, chemotherapy resistance, immune escape and postoperative recurrence of HCC [5].
Accumulating evidence suggests that lysophosphatidylcholine acyltransferase 1 (LPCAT1) is highly expressed and correlates with poor prognosis in the development of several types of cancer, including renal cancer, oral cancer, gastric cancer, colorectal cancer, prostate cancer and breast cancer [6–12]. LPCAT1 is mainly expressed in lung alveolar type II cells and has a crucial effect on pulmonary surfactant biosynthesis to support lung function [13]. LPCAT1 acts as a key enzyme in the phospholipid remodeling pathway [14] and catalyzes the conversion of lysophosphatidylcholine (LPC) into phosphatidylcholine (PC) [13]. A study published in 2013 stated that LPCAT1 is highly expressed in HCC and regulates hepatoma progression by altering phospholipid composition [15]. However, the report did not focus on the mechanism of metastasis and paid no attention to the effect of LPCAT1 on the biological behavior of HCC in vivo.
In this study, we further extended knowledge regarding the effects of LPCAT1 overexpression on HCC cell proliferation and invasion as well as pulmonary metastasis by establishing a spontaneous lung metastasis model in nude mice. Moreover, we observed that the expression pattern of EMT markers changed in response to overexpression or knockdown of LPCAT1 in HCC cells. Therefore, our data suggest that LPCAT1 contributes to the EMT process via the Wnt/β-catenin signaling pathway and promotes lung metastasis of HCC.
Materials and methodsClinical specimensThe collection of patient-derived specimens was in accordance with the Declaration of Helsinki and was approved by the institutional review board of Shanghai General Hospital. All HCC tissues and adjacent normal tissues were obtained from HCC patients at Shanghai General Hospital after surgery and immediately stored in liquid nitrogen. All patients provided written informed consent. This study was approved by the ethics committee of Shanghai Jiao Tong University, School of Medicine. Before undergoing surgery, none of the patients underwent any antitumor treatment, including hepatectomy, chemotherapy and radiotherapy.
Reagents and antibodiesAntibodies against β-catenin (#8480), phospho-β-catenin (Ser33/37/Thr41) (#9561), GSK-3β (#12,456), matrix metalloproteinase 7 (MMP-7) (#3801), T cell factor 1 and 7 (TCF1/TCF7) (#2203), E-cadherin (#3195), N-cadherin (#13,116), and vimentin (#5741) were obtained from Cell Signaling Technology (USA). Antibodies targeting LPCAT1 (ab214034) were purchased from Abcam (UK). Anti-β-actin (60,008–1-Ig) was purchased from Proteintech (Wuhan, China). D-Luciferin (E1605) was purchased from Promega (USA).
Cell lines and cell culture293T cells and the human HCC cell lines Hep3B and Huh7 were obtained from Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). The HCC cell line HCCLM3 was kindly provided by the Liver Cancer Institute of Zhongshan Hospital, Fudan University (Shanghai, China). 293T cells were cultured in DMEM supplemented with 5% fetal bovine serum (FBS, Gibco, USA); HCCLM3 and Huh7 cells were cultured in DMEM supplemented with 10% FBS (BI, SA) and 1% penicillin and streptomycin; and Hep3B cells were cultured in MEM supplemented with 10% FBS (BI, SA), penicillin and streptomycin. All cells were incubated in a humidified environment at 37 °C with 5% CO2.
Western blottingCell and tissue lysates were extracted using RIPA buffer (Biotechwell, Shanghai, China). Tissue and cell lysates were electrophoresed though SDS-PAGE gels (8%–12%; the concentration of the gels was chosen according to the protein molecular weight) and then transferred to PVDF membranes (Millipore, MA), which were blocked with 5% nonfat milk in TBST buffer for 60 minutes at room temperature (RT). Then, the membranes were incubated at 4 °C overnight with primary antibodies followed by incubation with secondary antibodies at RT for 1 hour the next day. The protein bands were detected using ECL reagent kits (Millipore, MA).
Transfection and generation of stable cell linesTo knock down LPCAT1 expression in Huh7 and HCCLM3 cells, plasmids containing pLKO.1 Vector or short hairpin RNA (shRNA) sequences targeting LPCAT1 were cotransfected with pMD2.G and psPAX2 into 293T cells to establish lentivirus particles according to the manufacturer's instructions. To construct LPCAT1-overexpressing Hep3B and Huh7 cell lines, plasmids containing the Flag-tagged coding sequence of LPCAT1 or pLenti-CMV-Puro were transfected into 293T cells as described above. Huh7, Hep3B and HCCLM3 cells were infected with lentiviruses containing empty vector, pLenti-CMV-Puro-LPCAT1, shLPCAT1 or scrambled shRNA to generate stably transduced cell lines, which were selected with puromycin (all plasmids were obtained from Obio Technology, Shanghai, China).
Cell migration and invasion assayIn vitro migration and invasion were evaluated using a Transwell 24-well chamber (Corning, USA) with membranes containing 8.0-μm pores. Matrigel (BD Biosciences, USA) was coated on the membranes in the invasion assay. Stably transduced Huh7, HCCLM3 and Hep3B cells in 200 μl of serum-free DMEM were seeded in the upper chamber, and 600 μl of medium supplemented with 10% FBS was added to the lower chamber. After 18–24 hours of incubation, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 15 minutes. Cells were counted in three random × 200 magnification fields per membrane. Three replicates per condition were used in each experiment, and the experiments were repeated in triplicate.
Hematoxylin and eosin (H&E) stainingFormalin-fixed, paraffin-embedded tissues were sliced into sections onto slides. After they were dewaxed with xylene, the slides were rehydrated in a series of graded ethanol solutions. Following immersion in hematoxylin staining solution, the sections were sequentially immersed in 1% hydrochloric acid and 0.5% eosin staining solution. Finally, the sections were dehydrated in a series of graded ethanol solutions, cleared with xylene and mounted. Observation and imaging were carried out under a microscope.
In vivo tumor growth and metastasis assaysAnimal experiments were approved by the Animal Ethics Committee of Shanghai General Hospital in accordance with EU Directive 2010/63/EU. Twenty 4-week-old male BALB/c nude mice were divided randomly into four groups (5 mice per group). For the subcutaneous tumor growth assay, Huh7 cells (3.5 × 106/100 μl PBS) overexpressing LPCAT1 or control vector were subcutaneously injected into the inguinal canal of nude mice. The tumor volume (volume=length*width2*1/2) was recorded every 2 days until its diameter exceeded 1.5 cm. For the metastasis experiments, cells were injected via tail vein, and tumor-bearing mice were subjected to in vivo imaging with an in vivo imaging system (IVIS) (Caliper IVIS Lumina, USA) before they were sacrificed. Immediately after excision, all the tumors were weighed, photographed, and fixed with formalin.
Statistical analysisStatistical analysis was performed with SPSS software (25.0). The independent experiments were repeated in triplicate for each assay. The results are presented as the mean ± standard deviation (SD). The Wilcoxon signed-rank test or Student's t-test was used to evaluate comparisons of measurable variants between groups. Values of P<0.05 were considered statistically significant. (*P<0.05, **P<0.01 and ***P<0.001).
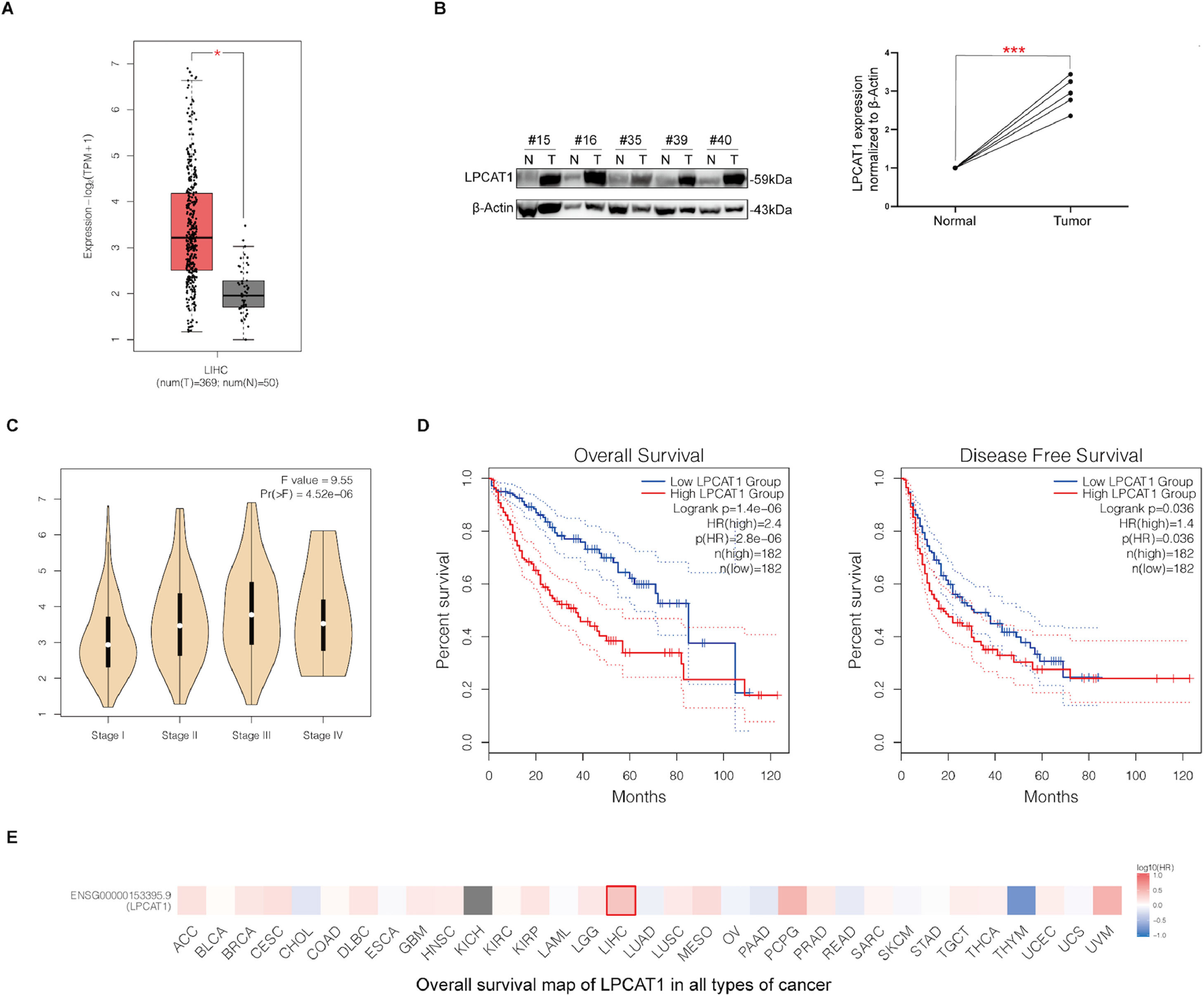
ResultsLPCAT1 is highly expressed in HCC tissues and indicates poor prognosis in HCC patientsWe first analyzed a public dataset from The Cancer Genome Atlas (TCGA) database and found that the LPCAT1 mRNA expression level was much higher in HCC tissues than in normal liver tissues (P < 0.01) (Fig. 1A). The western blotting results also confirmed that LPCAT1 protein levels were obviously upregulated in tissues of HCC patients compared with their matched surrounding normal tissues (Fig. 1B). Moreover, LPCAT1 expression was higher in HCC patients with advanced pathological stages than those with early pathological stages based on the 8th edition of the American Joint Committee on Cancer (AJCC) Staging System (Fig. 1C). Subsequently, we explored the relationship between LPCAT1 and both OS and disease-free survival (DFS) by conducting a Kaplan-Meier survival analysis with log-rank testing from 419 cases in the TCGA database. The graphs show that patients with tumors exhibiting high LPCAT1 expression had lower OS and DFS than those with tumors exhibiting low LPCAT1 expression (both P < 0.01) (Fig. 1D). The OS map of LPCAT1 in all types of cancer obtained from the TCGA database highlights its clinical significance with respect to the hazard ratio (HR) (Fig. 1E). In summary, the results above validate the prognostic value of LPCAT1 in HCC.
 LPCAT1 mRNA expression levels in clinical specimens from the TCGA database. (B) Western blotting analysis of LPCAT1 protein expression in five paired specimens. T: HCC tissues; N: adjacent normal tissues. β-Actin was used as the loading control. (C) The correlations between LPCAT1 expression levels and clinical tumor stage. (D) OS and DFS of HCC patients in the TCGA cohort with high or low LPCAT1 expression were estimated by Kaplan-Meier analysis and compared with the log-rank test. (E) OS map of LPCAT1 in all types of cancer showing its significance of LIHC. Statistical data in panels A and B are described as the mean ± SD. Error bars represent the SD. Asterisks indicate statistical significance: *P < 0.05, **P < 0.01 and ***P < 0.001 vs. normal tissues. The data in panels A, C, D and E were obtained from the TCGA database and analyzed by GEPIA (Gene Expression Profiling Interactive Analysis) (cancer-pku.cn). (Abbreviations: TCGA, The Cancer Genome Atlas; SD, standard deviation; OS, overall survival; DFS, disease-free survival; LIHC, liver hepatocellular carcinoma; HR, hazard ratio).")
(A) LPCAT1 mRNA expression levels in clinical specimens from the TCGA database. (B) Western blotting analysis of LPCAT1 protein expression in five paired specimens. T: HCC tissues; N: adjacent normal tissues. β-Actin was used as the loading control. (C) The correlations between LPCAT1 expression levels and clinical tumor stage. (D) OS and DFS of HCC patients in the TCGA cohort with high or low LPCAT1 expression were estimated by Kaplan-Meier analysis and compared with the log-rank test. (E) OS map of LPCAT1 in all types of cancer showing its significance of LIHC.
Statistical data in panels A and B are described as the mean ± SD. Error bars represent the SD. Asterisks indicate statistical significance: *P < 0.05, **P < 0.01 and ***P < 0.001 vs. normal tissues. The data in panels A, C, D and E were obtained from the TCGA database and analyzed by GEPIA (Gene Expression Profiling Interactive Analysis) (cancer-pku.cn). (Abbreviations: TCGA, The Cancer Genome Atlas; SD, standard deviation; OS, overall survival; DFS, disease-free survival; LIHC, liver hepatocellular carcinoma; HR, hazard ratio).
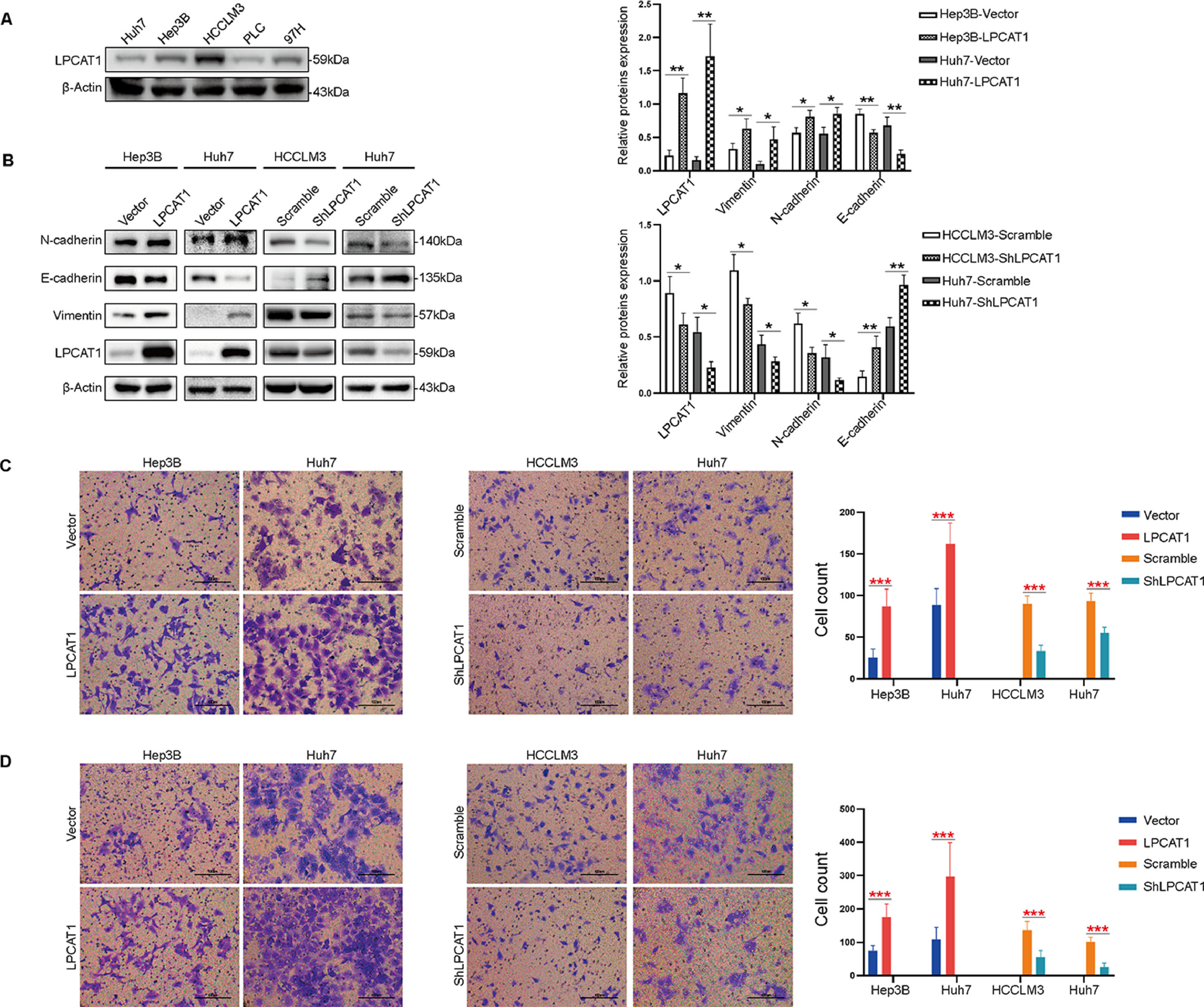
To confirm the hypothesis that LPCAT1 mainly affects invasion and metastasis in HCC, we first compared LPCAT1 levels in five common human HCC cell lines (Fig. 2A). The expression level of LPCAT1 was highest in HCCLM3 cells, which are characterized by their strong ability to spontaneously undergo pulmonary metastasis compared to the other HCC cell lines. In this study, we selected the most efficient shRNA from several different shRNA constructs to knock down LPCAT1 expression in Huh7 and LM3 cells (named Huh7-shLPCAT1 and LM3-shLPCAT1, respectively) for further experiments. Huh7 and Hep3B cells were also transduced with LPCAT1-overexpressing plasmids (named Hep3B-LPCAT1 and Huh7-LPCAT1, respectively) as described previously in the methods. Cells that served as negative controls for LPCAT1 overexpression and knockdown were named Hep3B-Vector, Huh7-vector, LM3-scramble and Huh7-scramble, respectively. The western blotting results verified the effectiveness of LPCAT1 knockdown or overexpression in the corresponding cells (Fig. 2B).
 LPCAT1 expression was assessed using western blotting assays in five HCC cell lines. β-Actin was used as a loading control. (B) Western blotting assays were used to detect the expression of EMT markers. (C,D) Transwell assays were performed to evaluate the in vitro migratory and invasive abilities of HCC cells with ectopic overexpression of knockdown of LPCAT1. Representative images are shown. 200X magnification; scale bars: 100 µm. Data are shown as the mean values±SD of at least three independent experiments. The statistical analysis compared the experimental and control groups with the unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001. (Abbreviations: TCGA, The Cancer Genome Atlas; SD, standard deviation; EMT, epithelial-mesenchymal transition).")
LPCAT1 promotes HCC cell migration and invasion by inducing EMT in vitro. (A) LPCAT1 expression was assessed using western blotting assays in five HCC cell lines. β-Actin was used as a loading control. (B) Western blotting assays were used to detect the expression of EMT markers. (C,D) Transwell assays were performed to evaluate the in vitro migratory and invasive abilities of HCC cells with ectopic overexpression of knockdown of LPCAT1. Representative images are shown. 200X magnification; scale bars: 100 µm.
Data are shown as the mean values±SD of at least three independent experiments. The statistical analysis compared the experimental and control groups with the unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001. (Abbreviations: TCGA, The Cancer Genome Atlas; SD, standard deviation; EMT, epithelial-mesenchymal transition).
We next assessed the expression pattern of EMT markers in the stably transfected cell lines mentioned above. Western blot analysis revealed that the protein levels of EMT markers dramatically changed after 24 hours of serum starvation. In Huh7 and LM3 cells with stable LPCAT1 knockdown, we observed higher E-cadherin levels than those in the control cells, and cells with stable LPCAT1 knockdown showed lower N-cadherin levels than those in the control cells. Meanwhile, we also found that the expression of the mesenchymal markers N-cadherin and vimentin were reduced in cells with of LPCAT1 knockdown and elevated in cells with LPCAT1 overexpression (**P < 0.01 for all) (Fig. 2B).
Moreover, we explored the invasive and metastatic capacities of the stably transfected cell lines. Transwell assays confirmed that the migration and invasion of cells with LPCAT1 overexpression significantly increased, whereas LPCAT1 knockdown inhibited cell migration and invasion in Huh7, Hep3B and HCCLM3 cell lines (***P < 0.001) (Fig. 2C,D).
In summary, these data demonstrate that LPCAT1 enhances HCC cell migration, invasion and EMT in vitro, hence we speculate EMT process might be the mechanism by which LPCAT1 contributes to HCC invasion and metastasis.
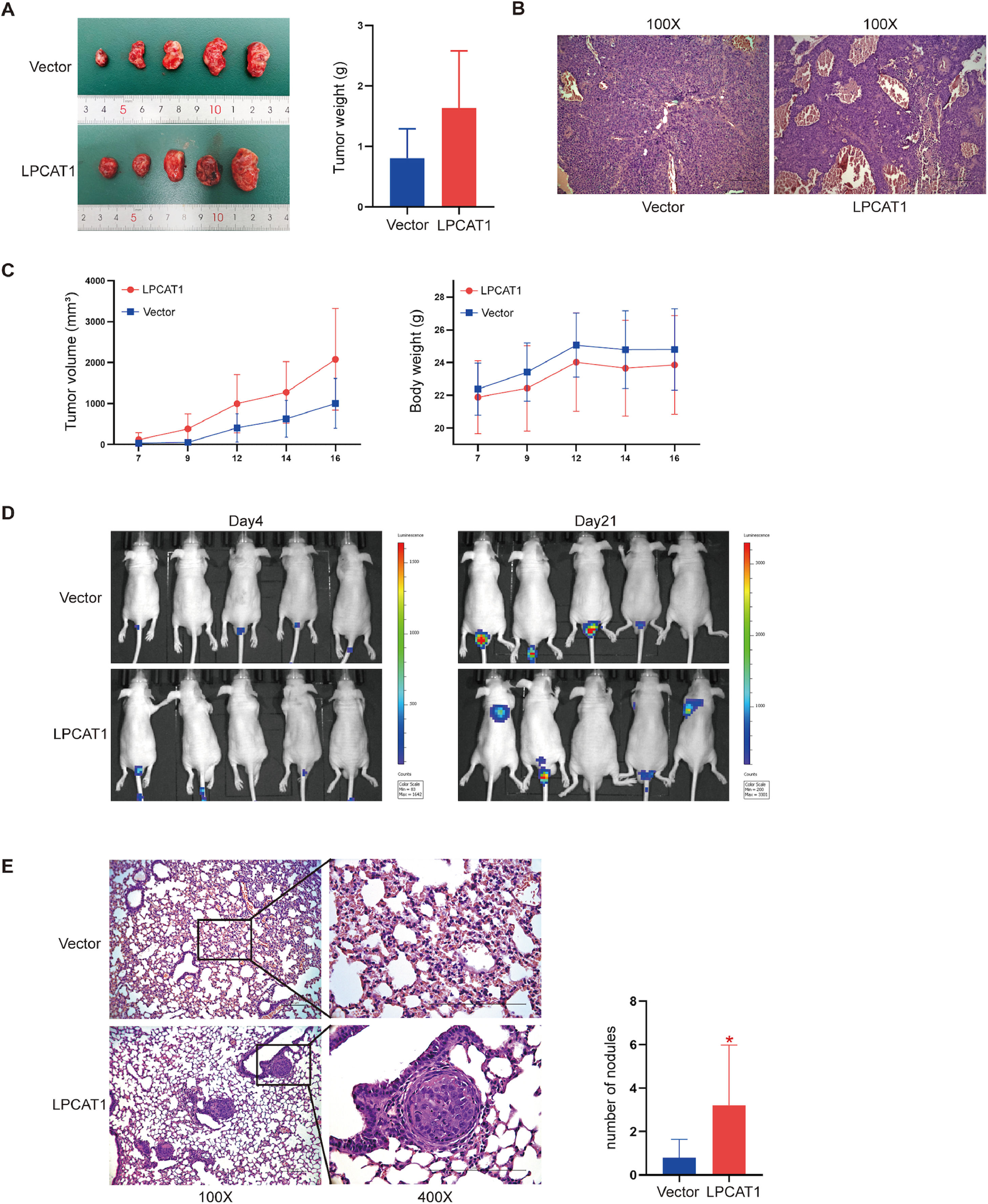
LPCAT1 overexpression accelerates HCC lung metastases in vivoTo investigate the tumorigenic effects of LPCAT1 on HCC in vivo, a subcutaneous cell-derived xenograft model was established using Huh7-LPCAT1 and Huh7-vector cells. We found that the mean tumor volume and tumor weight in the Huh7-LPCAT1 group were larger than those in the vector control group (Fig. 3A,C); unfortunately, there was no significant difference (both P>0.05). There was also no significant difference in the average total body weight between the two groups. H&E staining of subcutaneous tumors showed more vascular sinuses in the Huh7-LPCAT1 group (Fig. 3B). We used IVIS each week starting on postinjection day 4 to monitor and quantify metastatic progress in mice subjected to tail vein injection. Three weeks after the first administration, in vivo imaging was performed to identify metastatic lesions throughout the whole body (Fig. 3D). The results showed that the LPCAT1-overexpressing group had a higher metastatic rate than did the control group (lung: 40% vs. 0%). Using H&E staining, we further confirmed the results from the in vivo imaging, in which mice in the Huh7-LPCAT1 group developed more visible metastatic nodules, whereas the lungs of mice in the control groups showed normal tissue or tiny nodules (Fig. 3E). Similarly, H&E staining analysis of lung metastases showed that mice in the Huh7-LPCAT1 group had a higher pulmonary metastatic nodules rate than mice in the LPCAT1-vector group (Fig. 3E).
 Macroscopic images of subcutaneous tumors derived from Huh7-vector cells and Huh7-LPCAT1 cells were obtained on postinjection day 16. The tumor weight of xenografts was quantified after excision. The total body weight of nude mice and the total tumor volume of the xenografts were measured every two days (n=5 for each group). (B) Representative H&E-stained sections of xenograft tumors. (100X magnification; scale bars: 100 µm) (C) Metastatic progression was recorded and quantified with IVIS each week after tail vein injection. Representative bioluminescent images of the metastatic tumors are shown. (n=5 for each group) (D) Representative H&E staining images and statistical analysis of pulmonary metastatic nodules in tumors. (100X and 400X magnification; scale bars: 100 μm) (n=5 for each group; *P<0.05 vs. vector group) Each experiment was repeated at least three times. Error bars represent the SD. Statistical data in tumor formation assays and lung metastatic nodules are described as the means±SD. (Abbreviations: H&E, hematoxylin-eosin; IVIS, in vivo imaging system; SD, standard deviation).")
LPCAT1 overexpression accelerates HCC lung metastasis in vivo. (A) Macroscopic images of subcutaneous tumors derived from Huh7-vector cells and Huh7-LPCAT1 cells were obtained on postinjection day 16. The tumor weight of xenografts was quantified after excision. The total body weight of nude mice and the total tumor volume of the xenografts were measured every two days (n=5 for each group). (B) Representative H&E-stained sections of xenograft tumors. (100X magnification; scale bars: 100 µm) (C) Metastatic progression was recorded and quantified with IVIS each week after tail vein injection. Representative bioluminescent images of the metastatic tumors are shown. (n=5 for each group) (D) Representative H&E staining images and statistical analysis of pulmonary metastatic nodules in tumors. (100X and 400X magnification; scale bars: 100 μm) (n=5 for each group; *P<0.05 vs. vector group)
Each experiment was repeated at least three times. Error bars represent the SD. Statistical data in tumor formation assays and lung metastatic nodules are described as the means±SD. (Abbreviations: H&E, hematoxylin-eosin; IVIS, in vivo imaging system; SD, standard deviation).
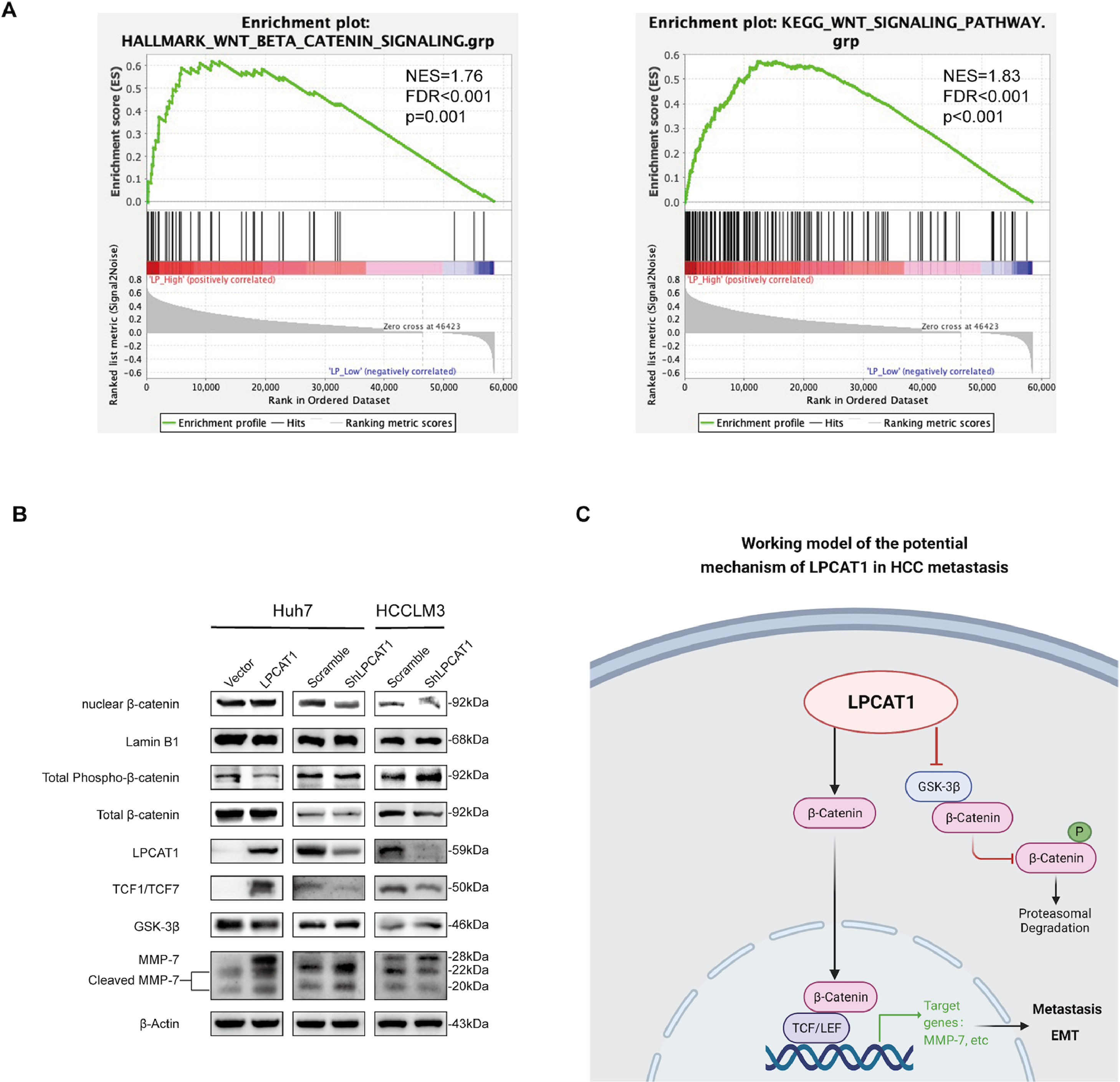
To systemically screen the potential pathways affected by LPCAT1 in HCC, we first analyzed differentially expressed genes in response to LPCAT1 by conducting Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis and gene set enrichment analysis (GSEA) from two different datasets from public databases (http://www.gsea-msigdb.org/gsea/msigdb/). Among the identified pathways, Wnt signaling was deemed one of the most significantly enriched pathways. There is evidence in the literature that Wnt is involved in HCC metastasis and invasion. GSEA plots indicated that the Wnt/β-catenin signaling pathway was significantly positively associated with LPCAT1 expression (NES=1.76, FDR q<0.001, p=0.001; NES=1.83, FDR q<0.001, p<0.001) (Fig. 4A). Hence, we detected the protein expression of the downstream effectors in the Wnt signaling pathway by western blotting analysis (Fig. 4B). To our astonishment, we observed that in Huh7-LPCAT1 cells, nuclear β-catenin, TCF1/TCF7 and activated MMP-7 levels were remarkably increased. Meanwhile, GSK-3β, cytoplasmic β-catenin and total phospho-β-catenin levels were decreased. By contrast, Huh7-shLPCAT1 cells exhibited the opposite expression patterns. In conclusion, the evidence indicates LPCAT1 might activate the Wnt/β-catenin signaling pathway to induce EMT based on the markedly altered expression of Wnt signaling effectors in response to overexpression or knockdown of LPCAT1 (Fig. 4C).
 The relationship between LPCAT1 expression and the Wnt/β-catenin signaling pathway was estimated by GSEA. The datasets were derived from the MSigDB database (http://www.gsea-msigdb.org/gsea/msigdb/). (B) The expression of key members of the Wnt/β-catenin signaling pathway and its downstream effectors was analyzed using western blot. (C) Schematic model of the function and potential mechanism of LPCAT1 in HCC metastasis was created with BioRender.com. (Abbreviations: GSEA, gene set enrichment analysis).")
LPCAT1 activates the Wnt/β-catenin signaling pathway in HCC cells. (A) The relationship between LPCAT1 expression and the Wnt/β-catenin signaling pathway was estimated by GSEA. The datasets were derived from the MSigDB database (http://www.gsea-msigdb.org/gsea/msigdb/). (B) The expression of key members of the Wnt/β-catenin signaling pathway and its downstream effectors was analyzed using western blot. (C) Schematic model of the function and potential mechanism of LPCAT1 in HCC metastasis was created with BioRender.com.
(Abbreviations: GSEA, gene set enrichment analysis).
LPCAT1 promotes metastasis and indicates poor prognosis in HCC. Studies have revealed that the expression of LPCAT1 is significantly increased in HCC, which fuels the progression of HCC in vitro [15–17]. However, it is still unclear how LPCAT1 mechanistically drives the process of HCC metastasis.
A study published in 2021 demonstrated that LPCAT1 regulates the progression of HCC by directly interacting with STAT117, which is more closely related to cell proliferation and cell cycle arrest of cancer cells instead of migration and invasion [18,19], even though it generally acts as a tumor suppressor [20]. Phospholipid levels in the cell membrane could affect membrane fluidity and facilitate the metastatic capacities of migration, adhesion and invasion of cancer cells [21,22], and a study utilizing imaging mass spectrometry (IMS) of clinical HCC tissues also corroborated the LPCAT1-mediated alterations in the phospholipid composition of the tumor cell membrane [15]. However, these studies did not focus on the direct relationship between the function of LPCAT1 as a lipid synthesis enzyme and the aggressive biological behaviors of HCC cells and therefore cannot clarify the association between LPCAT1 and HCC metastasis. Upon further investigation of the potential mechanisms of LPCAT1, we found for the first time that LPCAT1 contributes to HCC metastasis via EMT by activating the Wnt/β-catenin signaling pathway in vitro and promotes lung metastasis in vivo.
EMT is a complex program through which epithelial (E) cells transform into mesenchymal (M) cells, characterized by the loss of adherens junctions and the gain of metastatic dissemination. Conventional EMT has been described as the downregulation of E marker epithelial (E)-cadherin and the upregulation of the M marker neural (N)-cadherin, which is typically defined by a so-called “cadherin switch” [23]. During a complete EMT progress in embryonic development, epithelial-derived cells lose all epithelial phenotype vestiges and acquire a fully mesenchymal expression traits [24]. Conversely, cancer cells originating from epithelial cancers exhibit both epithelial and mesenchymal phenotype during EMT [23,24]. As shown in Fig. 2B, HCC cells undergone a typical cadherin switch in which the expression of E-cadherin and N-cadherin changed oppositely after forced expression of LPCAT1. Several recent studies suggest that EMT is a stepwise program rather than an impartible transition; hence, ‘incomplete’ or partial EMT is sufficient for tumor cell clusters to undergo migration and metastasis [24,25]. Carcinoma cells residing in various phenotypes along the epithelial-mesenchymal spectrum are correlated with more malignant phenotypes, such as metastasis, invasiveness and tumor formation, than cancer cells in a completely epithelial or mesenchymal state [24,25]. This conclusion can fully explain the western blotting results of Fig. 2B in that the trend of changes in the expression pattern of EMT markers is not significantly correlated with a complete EMT process in general, as we speculated. In other words, it is not necessary for all the typical markers, including E-cadherin, N-cadherin and vimentin, to observably change, as EMT is not an all-or-none switch. In conclusion, EMT is a much more flexible mechanism than initially thought.
The Wnt/β-catenin signaling pathway is considered to drive EMT in HCC since mutations of β-catenin were reported to occur in 13–41% of patients with hepatitis-induced HCC [26]. The WNT signaling pathways, including but not limited to the canonical WNT pathway, the noncanonical WNT-calcium pathway, and the noncanonical planar cell polarity WNT pathway, have been reported to participate in the EMT program [24,25,27,28]. As a key activator of EMT, the canonical WNT signaling pathway has been considered the foremost cascade among WNT signaling pathways [29]. β-Catenin, the core protein of the canonical WNT signaling pathway, can directly trigger migration by inducing the expression of EMT transcription factors (EMT-TFs) [30]. In turn, EMT-TFs can amplify the effect of the canonical WNT pathway by accelerating the transcription of β-catenin [24,31,32]. Conversely, E-cadherin is deemed to be negatively correlated with the canonical WNT signaling pathway, which promotes the displacement and translocation of β-catenin from adherens junctions on the cell membrane to the nucleus [33,34], where it can bind to TCF/LEF transcription factor family members to activate the transcription of downstream genes that initiate the EMT program [26]. Reduced E-cadherin expression, along with increased nuclear β-catenin expression, is observed in HCC and has been considered a positive marker of metastatic capacity and an independent prognostic factor of HCC [28,35]. Our experiment confirmed that MMP-7, as one of the most important downstream effectors activated by the Wnt/β-catenin signaling pathway, is accompanied by increased expression of LPCAT1 in HCC cells. Accumulating studies have demonstrated that MMP-7 can orchestrate cross-vascular invasion and migration by degrading the extracellular matrix (ECM) to promote metastasis and recolonization of tumor cells [36,37], which corresponds to the results of Fig. 1C showing that LPCAT1 indicates advanced tumor staging.
Considering the effect of LPCAT1 on phospholipid metabolism and its close association with the Wnt/β-catenin signaling pathway, we hypothesized that LPCAT1 promotes WNT signaling by affecting the phospholipid composition of the cell membrane, which will be specifically explored in our future studies.
There are several limitations to this study. The comparison of tumor volume and tumor weight of our xenograft model showed no significance between the experimental and control groups in Fig. 3A, even though the tumors from the LPCAT1-overexpressing group appeared to be larger in general. However, the negative result of significantly different tumor growth could be indicative that LPCAT1 promotes pulmonary metastasis. However, due to financial constraints, the expression of EMT-TFs such as Zeb1, Snail and Twist were not analyzed to further verify the internal regulatory network of EMT.
ConclusionsIdentification of specific genetic alterations and biomarkers related to HCC may facilitate earlier diagnosis and better treatment. Our study found for the first time that LPCAT1 enhanced the migration and invasion abilities of HCC cells via EMT and contributed to the lung metastasis of HCC. Thus, LPCAT1 could serve as a valuable prognostic biomarker and therapeutic target for patients with HCC.
FundingThis work was supported by the National Natural Science Foundation of China [Grant No.81270556]; the Natural Science Foundation of Shanghai [Grant No.19ZR1441300]; and the SJTU Interdisciplinary Program [Grant No. YG2021QN80].
Authors’ contributionsSL, YB, and SX conceived and designed this study. SL, GP, QC performed the experiments and analyzed the data. SL and GP drafted the manuscript. SL, DWT, and ZL prepared the figures. CWY, LZY, YB, and SX revised the manuscript. All authors read and approved the submitted version.
Ethical approvalThis study was approved by the Ethics Committee of Shanghai General Hospital of Shanghai Jiao Tong University School of Medicine.
Written informed consent was obtained from all participants.
We sincerely acknowledge TCGA and GEPIA databases for providing their platforms and contributors for uploading their datasets.
recomendados



![]()
