



Lysosomal acid lipase deficiency is a poorly diagnosed genetic disorder, leading to accumulation of cholesterol esters and triglycerides in the liver, with progression to chronic liver disease, dyslipidemia, and cardiovascular complications. Lack of awareness on diagnosis of this condition may hamper specific treatment, which consists on enzymatic replacement. It may prevent the progression of liver disease and its complications. We describe the case of a 53-year-old Brazilian man who was referred to our center due to the diagnosis of liver cirrhosis of unknown etiology. He was asymptomatic and had normal body mass index. He had dyslipidemia, and family history of myocardial infarction and stroke. Abdominal imaging tests showed liver cirrhosis features and the presence of intrahepatic calcifications. Initial investigation of the etiology of the liver disease was not elucidated, but liver biopsy showed microgoticular steatosis and cholesterol esters deposits in Kuppfer cells. The dosage of serum lysosomal acid lipase was undetectable and we found the presence of a rare homozygous mutation in the gene associated with the lysosomal acid lipase deficiency, (allele c.386A > G homozygous p.H129R).
Lysosomal acid lipase deficiency (LAL-D) is a multisystem autosomal recessive genetic disease caused by mutations in the LIPA gene, which encodes the enzyme lysosomal acid lipase (LAL), responsible for hydrolyzing the cholesteryl esters and triglycerides within low density lipoprotein (LDL) particles into free cholesterol and free fatty acids.1 LAL-D is an under-recognized condition, frequently misdiagnosed, and the time to accurate diagnosis of LAL-D is usually delayed, leading to chronic liver disease, dyslipidemia and cardiovascular complications.2Many patients receive incorrect diagnoses, since the disease may simulate many other disorders as familial hypercholesterolaemia, familial combined hyperlipidaemia, non-alcoholic steatohepatitis, non-alcoholic fatty liver disease, chronic liver diseases or cryptogenic cirrhosis.1-3
LAL-D leads to accumulation of cholesteryl esters and triglycerides in the lysosomes of many tissues, including the liver, the spleen, the cardiovascular system and the macrophages throughout the body.1,2 The disease presents as two major phenotypes: a rapidly progressive form, called Wolman disease, which manifests in childhood, and a form that manifests later, also called cholesteryl ester storage disease (CESD).4 Liver damage with progression to fibrosis, cirrhosis and liver failure occurs in a large proportion of patients. Bernstein, et al. reported liver biopsy findings in 135 patients with cholesteryl ester storage disease: 112 of 135 showed microvesicular steatosis leading to fibrosis, micronodular cirrhosis, and liver failure in some patients.1 Extrahepatic manifestations included esophageal varices and accelerated atherosclerosis.1,2
There are few reports of LAL-D patients worldwide but the recognition of the disease is fundamental because there is a perspective of treatment that can considerably modify the natural history of the subjects.4,5 To our knowledge, this is the third case of LAL-D with allele c.386A > G homozygous p.H129R mutation.6
Case ReportA 53-year-old Caucasian man was referred to our center in 2005 with a chronic liver disease diagnosis that was based on the findings of an asymptomatic splenomegaly and thrombocytopenia that were detected in the previous year. On physical examination, hepatosplenomegaly with no signs of ascites or jaundice was noted. His body mass index was 23.8 kg/m2 and he had a family history of cardiovascular disease: his brother died at the age of 55 as a consequence of an acute myocardial infarction and his father, who had arterial hypertension, had a fatal stroke.
Initial laboratory tests showed liver enzyme elevation: aspartate aminotransferase (AST) was 91 IU/L [reference value (RV) < 50 IU/L], alanine aminotransferase (ALT) was 85 IU/L (RV < 50 IU/L), alkaline phosphatase was 131 IU/ L (RV < 129) and gamma glutamyl transferase (GGT) was 81 IU/L (RV < 40). LDL cholesterol (192 mg/dL; RV < 100 mg/dL), triglycerides (230 mg/dL; RV < 150 mg/dL) and HDL (high density lipoprotein) cholesterol (21 mg/dL; RV > 40 mg/dL) were also abnormal. He had normal liver function tests and was classified as Child-Pugh A.
Investigation was negative for autoimmune and infectious diseases and serum protein electrophoresis was normal. Abdominal ultrasound showed signs of chronic liver disease and punctate hyperechoic images with posterior acoustic shadows scattered by the liver parenchyma, suggestive of intrahepatic calcifications. There were splenomegaly and signs of portal hypertension.
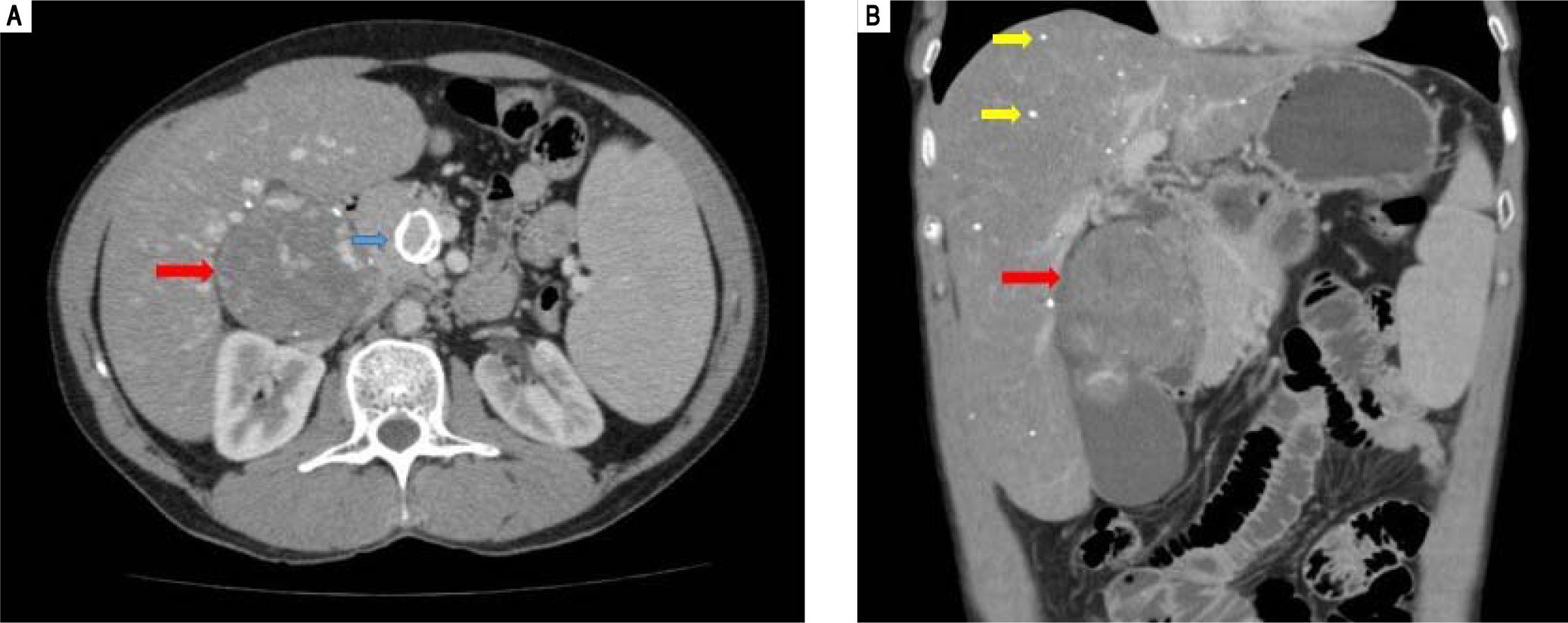
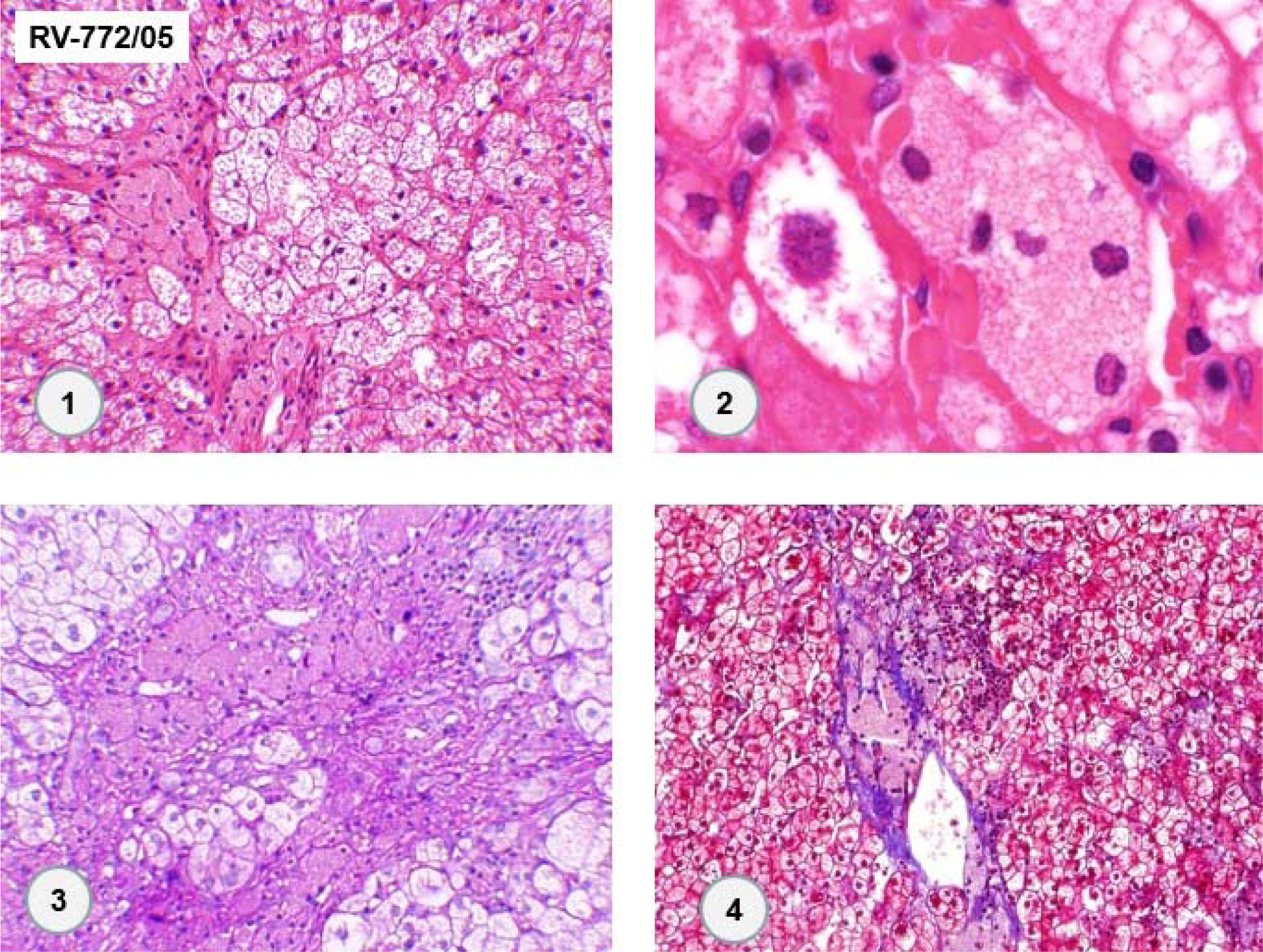
An abdominal computed tomography scan showed hepatomegaly with signs of fatty infiltration of the liver (steatosis), diffuse intrahepatic calcifications, splenomegaly and abdominal lymphadenomegaly. There was a 80 mm round image in the hepatic hilus, with punctuate calcifications and heterogeneous uptake of intravenous contrast (Figure 1). The lesion was surgically resected (as described latter), setting the diagnosis of right adrenal hemangioma. A liver biopsy was undertaken in 2005, showing chronic liver disease in multifocal transformation phase (liver cirrhosis) with extensive and diffuse microgoticular steatosis and abundant portal macrophages, suggestive of storage disease, but without a specific diagnosis (Figure 2).
, a calcified aneurysm in the superior mesenteric artery (blue arrow) and the presence of a rounded lesion, measuring 80 mm at its large diameter, with points of calcification and heterogeneous impregnation of the contrast (red arrows).")
Computed tomography of the abdomen. A. transverse. B. Coronal. There is hepatomegaly, splenomegaly, fatty liver infiltration with diffuse puntacte calcifications (yellow arrows), a calcified aneurysm in the superior mesenteric artery (blue arrow) and the presence of a rounded lesion, measuring 80 mm at its large diameter, with points of calcification and heterogeneous impregnation of the contrast (red arrows).
. (1. HE 400x. 2. HE 1,000x. 3. PAS 400x. 4. Masson-trichrome 100x).")
The patient had been followed as having crytogenic cirrhosis. For the evaluation of portal hypertension, an upper endoscopy performed in 2007 showed portal gastropathy and duodenal polyps, which on biopsy were confirmed as xanthomas. However, in the last endoscopy performed in 2015, we noted small esophageal varices, suggestive of progression of the liver disease.
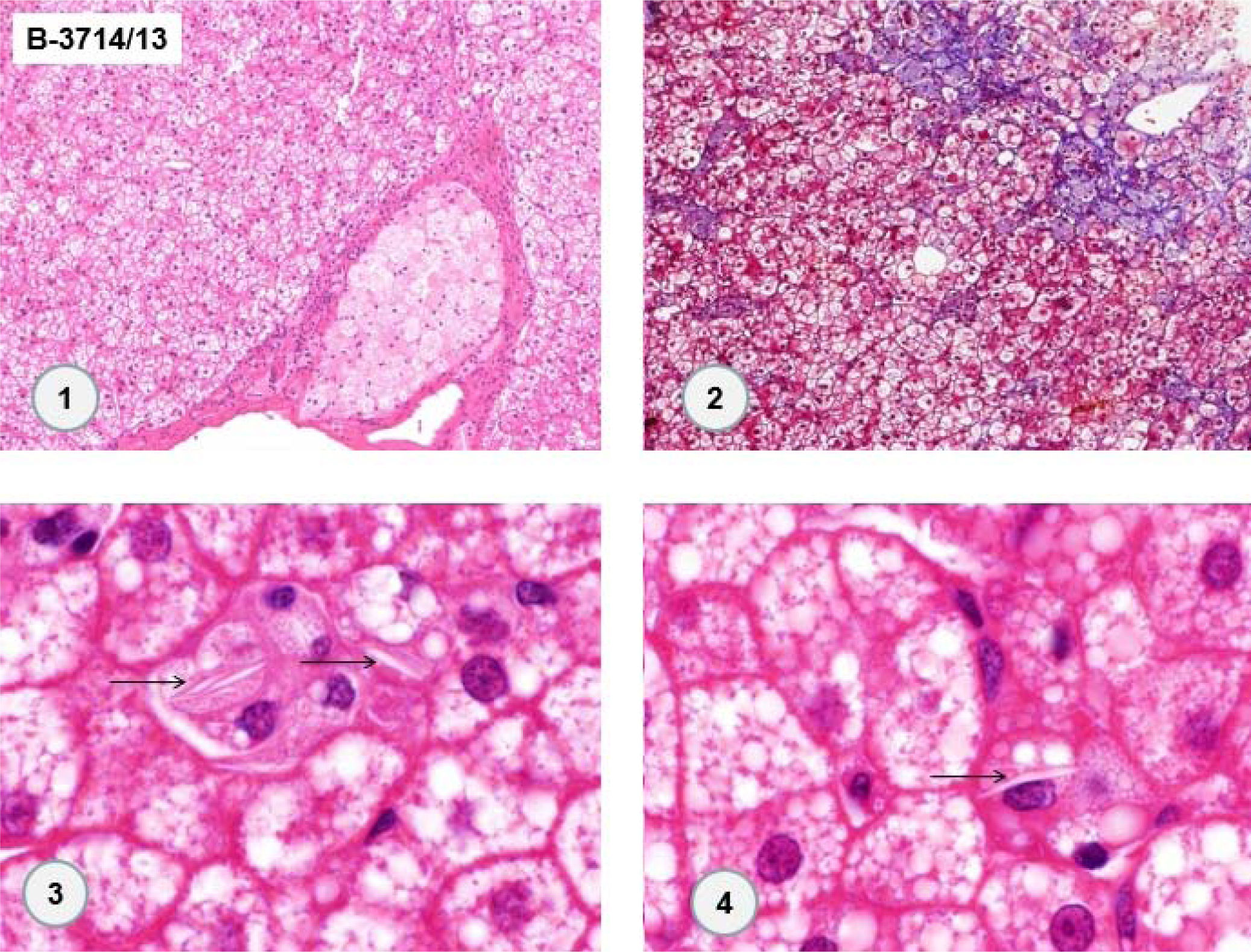
In 2013, the patient underwent surgery for resection of adrenal cavernous hemangioma (previously described) and cholecystectomy, and a new liver biopsy was performed during the surgery. Histological examination of the liver fragment (Figure 3) showed the presence of intense and diffuse microgoticular steatosis associated with macrophages containing focal deposits of cholesterol crystals was suggestive of lysosomal acid lipase deficiency.
 in some Kupffer cells. (1. HE 100x. 2. Masson-trichrome 100x. 3 and 4. HE 1,000x).")
The dosage of lysosomal acid lipase was undetectable using the blood spot test (at Seattle Children’s Hospital) and a rare pathogenic variant mutation was observed in homozygous for the LIPA gene (allele c.386A > G homozygous p.H129R), using Sanger dideoxy sequencing method (Ref Sequence NM_000235.3.), confirming the LAL-D diagnosis. The dosage of lysosomal acid lipase and LIPA gene sequencing were provided by Alexion Pharmaceuticals under our request. The patient was referred for cardiovascular evaluation.
DiscussionWe reported a case of a patient who had been followed for several years as having cryptogenic cirrhosis, with no specific treatment for his disease, and experienced progression of liver damage, reflected by the worsening in portal hypertension at the endoscopy surveillance.
The prevalence of cryptogenic cirrhosis ranges from 5% to 30% in large centers.7 Non-alcoholic steatohepatitis (NASH) is a possible cause of liver disease in many patients with cryptogenic cirrhosis, and collagen deposition toward advanced fibrosis can replace macroesteatosis and other histological features of NASH. In this context, the liver biopsy may be unable to confidently diagnose cirrhosis due to NASH. Moreover, there is a strong association between the presence of obesity, diabetes and cryptogenic cirrhosis.7,8 Identifying the etiology of liver disease is important to institute specific treatment when available, aiming to delay the progression of liver disease, to reduce the risk of complications, the need for liver transplantation and ultimately mortality.
Although the most common causes of chronic liver disease are generally researched and addressed, rare causes and related treatments are often forgotten, restricting the medical follow-up to the management of complications of cirrhosis, such as ascites, upper gastrointestinal hemorrhage, hepatic encephalopathy and hepatocellular carcinoma.7 LAL-D is part of the causes of poorly diagnosed liver disease, but its knowledge is important because it can lead to liver cirrhosis associated with early dyslipidemia and atherosclerosis. Another diagnostic challenge is that LAL-D can also mimic the clinical picture of non alcoholic fatty liver disease (NAFLD).9 We believe LAL-D must be considered in the work up evaluation of suspected NAFLD patients that lack features of the metabolic syndrome other than dyslipidemia, in an individualized approach, however further studies are needed to address this topic. Family screening is also advised after a LAL-D diagnosis.
Patients presenting LAL-D in infancy have the most rapidly progressive disease, developing signs and symptoms in the first weeks of life and rarely surviving beyond six months of age. Children and adults typically present with some combination of dyslipidemia, hepatomegaly, elevated transaminases, and microvesicular liver steatosis on histological evaluation. Elevated low-density lipoprotein cholesterol levels and decreased high-density lipoprotein cholesterol levels are common features, and cardiovascular disease may manifest as early as in childhood.2
LAL-D arises from mutations in the LIPA gene, which maps to chromosome 10q23.2. The most commonly inherited defect is the exon 8 splice site mutation, E8SJM (c.894G > A), which is found in more than half of all children and adults with LAL-D.2,10 Our patient had a different mutation in both alleles c.386A > G, p.H129R. And this is the third description of this mutation in homozygous.6 Ries, et al. reported a Swiss patient with one allele codifying p.H129R and the other described as c.64-?_428+?del (Ex2_4del).11 The c.386A > G LIPA gene mutation is associated with residual LAL activity,2 but in our patient, its presentation in homozygous may have resulted in absence of activity of this enzyme.
In a longitudinal analysis of 48 LAL-D patients assessed at a median of 12.8 years, hepatomegaly was the most common initial clinical finding (54% of the cases), and cholecystopathy occurred in 10% of them. Elevations of ALT were frequent in 92% of the cases, and of AST in 59%. Laboratory abnormality of GGT was no longer as relevant (observed in only 20% of the cases). LDL, total cholesterol and triglycerides averages were already high at the time of the first measurement in 64.4%, 62.5%, and 27.1% of the patients, respectively. Mean HDL cholesterol at the first dosage was altered in less than half (43.5%) of the patients. Major imaging findings were hepatomegaly (77%), splenomegaly (64%) and liver steatosis (51%). Adrenal calcification was observed in 3 cases. Liver biopsies performed in 31 patients showed: steatosis in 87% and fibrosis/cirrhosis in 64.5%. Liver transplantation was performed in 13 patients. Almost 40% of the patients in the study had a brother with LAL-D.12 Our patient had dyslipidemia, hepatomegaly, splenomegaly, and cirrhosis of unknown etiology. Liver biopsy findings and the important family history of cardiovascular disease were relevant for the suspicion of LAL-D. Despite reports in scientific meetings, we did not find publications associating intrahepatic calcifications with LAL-D.
Studies with low cholesterol/reduced saturated fat diets, HMG-CoA reductase inhibitors and vitamin E have been carried out, but low efficacy and unclear benefit in preventing cardiovascular and liver disease progression compromise these treatment strategies.13-15 Hematopoietic stem cell transplantation (a potentially curative treatment, as LAL activity could be restored from the donor cells) has also been evaluated, but varied reported outcomes, failure of engraftment, risks, and post-transplant complications have stimulated the search for additional treatments.16-19 Liver transplantation has also been attempted in both infants and adults with LAL-D in very limited case reports. In those with end-stage liver disease, they suggest good outcomes,20-23 however shortage organ availability, lack of specific criteria for transplantation, procedure associated risks and de novo graft liver disease concern limits its wide application.15
Enzyme replacement therapy has brought more promising results. In the first human study of the clinical effects and safety of the recombinant human LAL, sebelipase alfa, nine patients received four once-weekly infusions in a multicenter open label trial (LAL-CL01 Study). Enzyme replacement was well tolerated, with mostly mild adverse events unrelated to sebelipase alfa (nausea, diarrhea and headache).4 There was a rapid decrease in serum transaminases, with sustained results with long-term dosing, 52 weeks (LAL-CL04 Study), and accompanied by improvements in serum lipid profile.4,24
A multicenter randomized double blind placebo controlled phase III trial (LAL-CL02/ARISE Study) was carried out with 20 weeks of observation in 66 patients and an open label extended evaluation for 36 weeks. Sebelipase showed a significant reduction rate of ALT, AST and GGT in the treatment group compared with the placebo group (p < 0.001). The reduction of fatty liver content evaluated by magnetic resonance was greater in the treatment group (p < 0.001). There was also a significant improvement in the lipidogram in the sebelipase group compared to placebo, and it occurred independently of the use of hypolipidemic drugs. The open label period of the study (36 weeks) sustained findings of LDL and ALT improvement in the sebelipase group and showed improvement in the placebo group who started the medication in this period. Moreover, enzymatic replacement was associated with a reduction of 206.7% of the 10 year Framingham Risk Score estimation over 20 weeks of treatment, compared to placebo.25 These findings demonstrated that alpha sebelipase may improve markers of liver damage and fatty liver content and could change the natural course of this disease.5,26 Sebelipase was approved by the FDA and EMEA in 2015 for long-term replacement therapy in patients of all ages with LAL-D.27,28 In October 2017 the enzyme was also approved by the Brazilian Regulatory Agency - ANVISA.29 Close follow up of the patient after replacement therapy will be undertaken.
ConclusionsWe described a case of lysosomal acid lipase deficiency in a Brazilian man adjudicated as having cryptogenic cirrhosis for several years. LAL-D is an unusual etiology of liver disease and should be investigated in patients with dyslipidemia, especially low HDL cholesterol, persistently elevated aminotransferases or microgoticular hepatic steatosis of unknown cause. The diagnosis is important because there is a new therapeutic approach, which may prevent the progression of liver disease and its complications. However, further studies are needed to address these issues
Abbreviations- •
ALT: alanine aminotransferase.
- •
AST: aspartate aminotransferase.
- •
CESD: cholesteryl ester storage disease.
- •
GGT: gamma glutamyl transferase.
- •
HDL: high density lipoprotein.
- •
LAL: lysosomal acid lipase.
- •
LAL-D: lysosomal acid lipase deficiency.
- •
LDL: low density lipoprotein.
- •
NASH: non-alcoholic steatohepatitis.
There was no financial support for this study.
Conflict of InterestThe authors declares that there is no conflict of interest regarding the publication of this article.



