




Intrahepatic hypoxia may occur during the inflammatory and fibrotic processes that characterize several chronic liver diseases of viral and autoimmune origin. As a consequence, new vascular structures are formed to provide oxygen and nutrients. Angiogenesis involves a tightly regulated network of cellular and molecular mechanisms that result in the formation of functional vessels. Of particular importance are growth factors and molecules involved in matrix remodeling and cell migration, as weel as vessel maturation-related factors. In recent years a number of studies have investigated the expression and function of many pro-and antiangiogenic molecules in chronic liver diseases and liver regeneration. This review examines the potential pathogenic role of angiogenesis in the context of viral hepatitis, autoinmmune hepatitis, primary biliary cirrhosis and hepatocellular carcinoma.
Abbreviations
E-selectin, endothelial selectin
ECM, extracellular matrix
EC, endothelial cells
NO, nitric oxide
VEGF, vascular endothelial growht factor
aFGF and bFGF, acidic and basic fibroblast growth factors
HGF, hepatocyte growth factor
KDR, kinase insert domain receptor
Flt-1, fms-like tyrosine kinase receptor
CHC, chronic hepatitis C
HCV, hepatitis C virus
Ang, angiopoietin
ALT, alanine aminotransferase
PBC, primary biliary cirhosis
PSC, primary sclerosing cholangitis
AIH, autoimmune hepatitis
HCC, hepatocellular carcinoma
IntroductionAngiogenesis, the formation of new vascular structures from preexisting ones, occurs in several organs during multiple pathophysiological situations.1 It has become one of the most thoroughly investigated patophysiological phenomena in the last few years because of the key roles it plays in disease pathogenesis and its potential as a therapeutic target. Although mostly studied in relation to cancer, angiogenesis is also known to occur in pathologies characterized by chronic inflammation. Recent work from our group and others has demonstrated that chronic diseases of the liver do not represent an exception to this rule.
In 1995, our group2 studied by immunohistochemistry the intrahepatic expression pattern of vascular adhesion molecules (CD31, E-selectin, VCAM-1, cadherin 5 and endoglin) in viral chronic hepatitis. A novel and striking finding in this study was the detection of CD31+ and cadherin 5+ endothelial cells with microvessel morphology in the inflamed portal tracts from patients with viral chronic hepatitis. Furthermore, the expression pattern of both CD31 and cadherin 5 molecules on endothelial cells was always similar in enlarged portal tracts of these patients, acquiring a characteristic form of capillary tube formation. The assumption for an endothelial cell specificity was further reinforced, because cadherin 5 is a selective endothelial cell marker. It was also interesting to note that the presence of these portal microvessels was more important in those cases where portal tracts were more inflamed. By contrast, a weak immunoreactivity for CD31, cadherin 5, and endoglin was only found on sinusoidal endothelial cells and on portal vascular endothelium in normal liver tissue. This work gave important information about the potential pathogenetic role of angiogenesis in the context of liver disease.
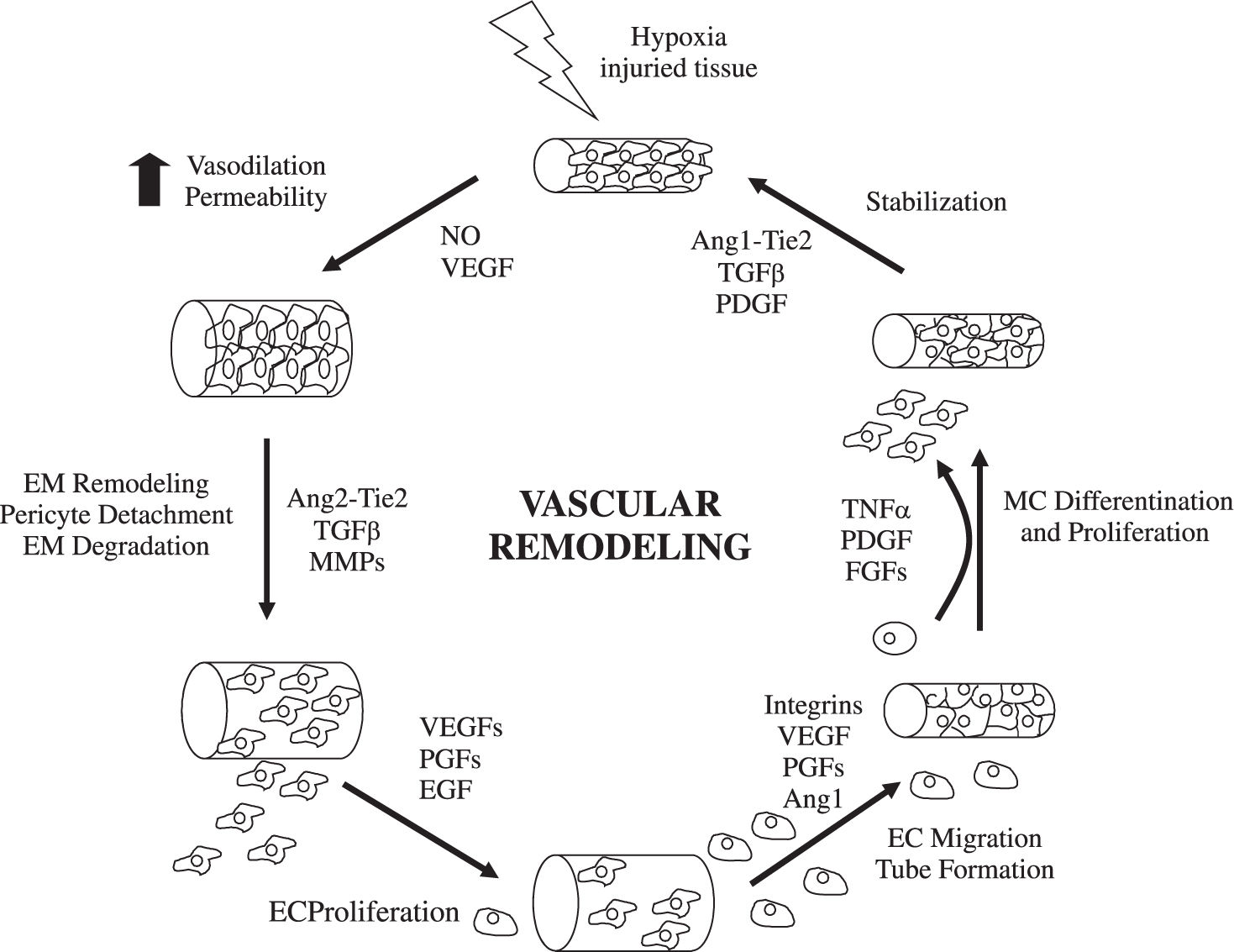
Mechanisms of angiogenesisHypoxia and inflammation are the main stimulous for angiogenesis. Hypoxia promotes the angiogenesis by signalling through hypoxia inducible transcription factors.1,3 During inflammation, vascular permeability is increased and monocytes, macrophages, platelets, mast cells, and other leukocytes (able to produce angiogenic cytokines and growth factors) are recruited under the attraction of chemokines4,5(Figure 1). The formation of new functional vessels involves different processes whose molecular effectors must be precisely regulated.6,7Medina et al1 reviewed the evidence for angiogenesis in chronic inflammatory liver diseases and analyzed the cellular and molecular mechanisms involved.

Endothelial budding is facilitated by vasodilation, loosening of interendothelial contacts, and leakiness of preexisting vessels, which allows extravasation of plasma proteins that, together with extracellular matrix components (ECM), lay down a provisional scaffold for migrating endothelial cells (EC). Nitric oxide (NO), whose angiogenic properties have been characterized,8 is the main factor responsible for vasodilation, whereas vascular endothelial growth factor (VEGF) increases vascular permeability. Next, the basement membrane (mainly collagen IV and laminin) and the ECM (collagen I and elastin) must be degraded to allow subsequent EC migration and proliferation. This is performed by specialized proteinases, including plasminogen activator. ECM proteolysis leads to the exposure of cryptic epitopes and release of ECM-embended factors that promote EC migration and proliferation.9,10
EC proliferate in response to growth factors secreted by EC or surrounding cells (including hepatic stellate cells, leukocytes, hepatocytes and Kupffer cells). The most thoroughly characterized is VEGF, a multifunctional protein that binds to 2 tyrosine kinase receptors: kinase insert domain receptor (KDR) and fms-like tyrosine kinase receptor (Flt-1).11 The VEGF promoter contains hypoxia-inducible factors-responsive elements. VEGF plays a crucial role in virtually all pathological situations in which angiogenesis occurs, and the therapeutic use of strategies aimed at blocking its mode of action is currently under evaluation.12 In addition, EC proliferation may be stimulated by other growth factors, such as acidic and basic fibroblast growth factors (aFGF and bFGF); hepatocyte growth factor (HGF); and transforming growth factor (TGF).7,13
EC proliferate in an ordered manner that leads to the formation of a lumen.14,15 A structured 3-dimensional network of vessels of uniform size is then organized by carefully regulated mechanisms involving signaling pathways that determine branching, formation of basement membrane and ECM components, and cell migration and differentiation. For nascent vessels to mature, pericytes must be recruited, and a new basement membrane and ECM must be generated to provide structural stabilization.15 Physical forces and multiple molecules contribute to these processes.
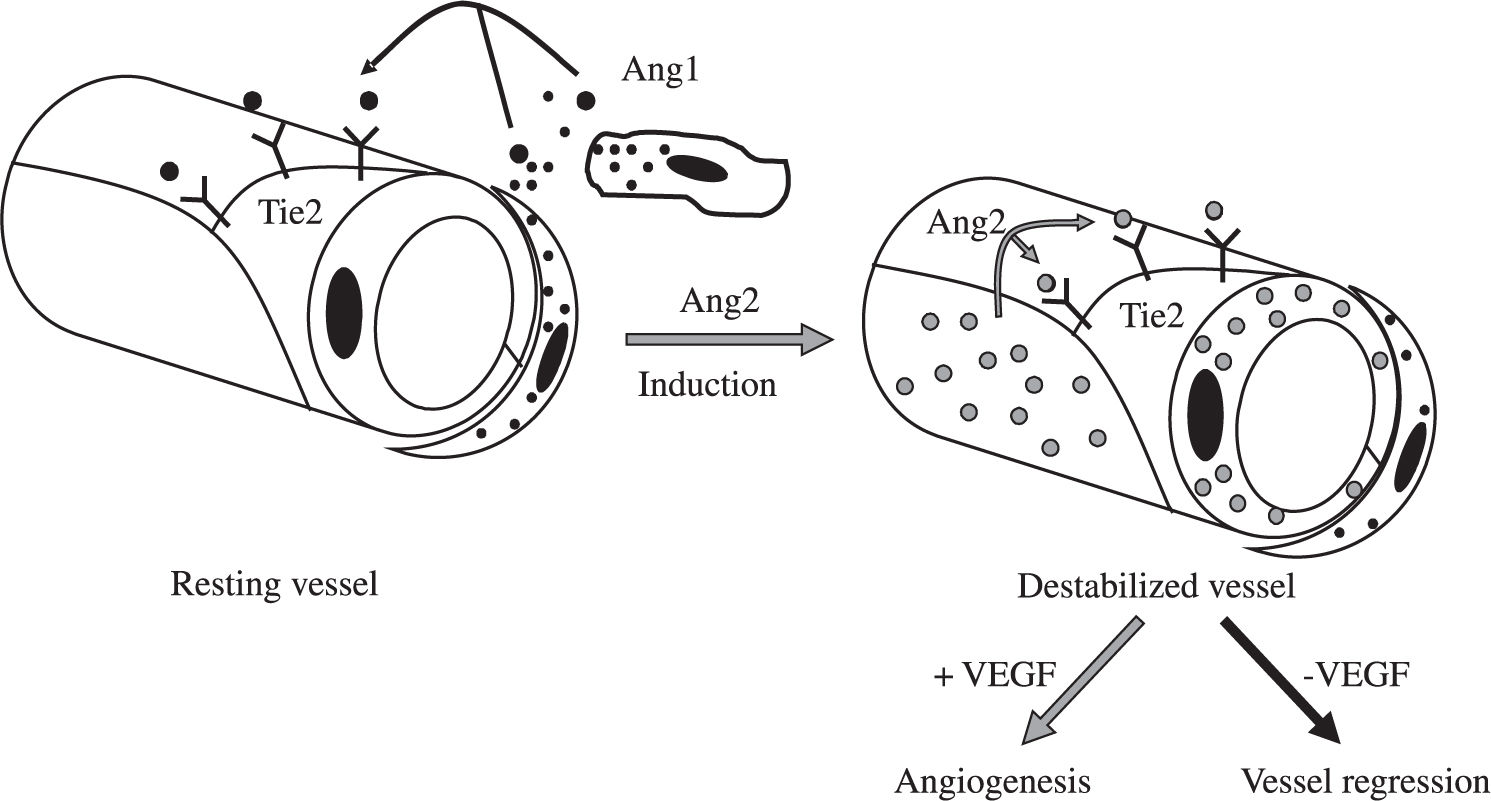
Besides induction of EC proliferation, effective angiogenesis also requires stabilization of the nascent vessels, establishment of interndothelial junctions and formation of a lumen.1 Angiopoietin 1 (ang-1) stabilizes neovessels by binding to the Tie-2 receptor, thereby affecting junctional molecules16 and facilitating communication between EC and mural cells.17 However, an excess of Ang-1 makes vessels too tight and inhibits sprouting. Ang-2 may exert opposing effects (Figure 2): in the absence of VEGF, Ang-2 acts as an antagonist of Ang-1, destabilizes vessels and causes EC death, leading to vessel regression, but it facilitates sprouting in the presence of VEGF.17

Most molecular mechanisms of angiogenesis are common to the liver and other organs. Physiological hepatic angiogenesis occurs during liver regeneration, leading to the formation of new functional sinusoids, whereas pathological angiogenesis occurs in fibrosis and is characterized by the appearance of capillarized vascular structures. Most chronic liver diseases are characterized by fibrosis and inflammation. During the fibrogenic process, an excessive amount of ECM is synthesized and accumulated. Fibrotic tissue offers resistance to blood flow and to the delivery of oxygen, thus becoming hypoxic. Stimulaton of hypoxia-inducible factors leads to an angiogenic switch, to the upregulation of proangiogenic factors, and to the formation of neovessels.2
Angiogenesis in chronic viral hepatitisAngiogenesis occurs in the liver of patients infected with hepatitis B or C virus.1,2 The pathophisiological significance of chronic viral hepatitis-associated angiogenesis is presently unclear; it has been proposed to exert a beneficial role by contributing to tissue repair and regeneration after liver damage.2 It has also been suggested to represent a risk factor for progression to hepatocellular carcinoma in patients with chronic hepatitis C.4
The molecular mechanisms involved in chronic viral hepatitis-associated angiogenesis have not been fully identified. Local production of NO as a result of the overexpression of inducible NO synthase in the livers of patients infected with hepatitis C or B virus may participate in the angiogenic response by inducing vasodilatation.18,19 VEGF and HGF, whose expression is increased during chronic viral hepatitis,20-22 may contribute to enhancing vascular permeability, as suggested by the effect described for these two growth factors. VEGF has been reported to induce NO-mediated vasodilation,11 most probably contributing to the progression of neoangiogenesis.2 HGF effects include a decrease and redistribution of VE-cadherin (which participates in intercellular contacts), and others linking molecules between VE-cadherin and the actin cytoskeleton. Additionally, viral proteins may also play a role in inducing a disruption of interendothelial junctions23 through mechanisms involving Src kinases, molecules required for vascular permeability during angiogenesis.24 In this way, Sanz-Cameno et al decribed for the first time the ability of hepatitis B virus (HVB), through HBx protein, to promote Ang-2 expression in liver tissue of chronically infected patients,25 and Lara-Pezzi et al found that matrix metalloproteinase 2 is also up-regulated by HBx protein.26 Binding between EC and ECM may be altered in the livers of chronic viral hepatitis patients.27 Integrin av• •3, expressed by activated EC, shows increased tissular expression in chronic hepatitis C (CHC).22 Interestingly, when EC are induced to migrate by HGF, integrin av• •3 accumumaltes at the leading edge,22 which represents the anchoring zone required for pulling the cellular body28(Table I).
Angiogenesis soluble factors with potential to be used as biomarkers.
| Factor | Receptor/s | Inhibitors | Effects | Process involved |
|---|---|---|---|---|
| VEGF family members | Flt-1 Flk-1/KDR | sFlt-1 | Increase vascular permeability by loosening intercellular contacts Regulate neovessel lumen formation Induce leucocyte adhesion | Sprouting/budding, vessel gowth 3-D organization |
| Ang-1 | Tie-2 | sTie-2 | Stabilizes intercellular contacts Inhibit permeability | Stabilization of nascent vessels |
| Ang-2 | Tie-2 | sTie-2 | In the presence of VEGF, it increases capillary diameter and remodels basal lamina In the absence of VEGF, it desestabilizes Vessels and causes EC death | Vessel regresion |
| aFGF, bFGF, | FGF-R1, FGF-R2 | Angiostatin | Induce EC proliferation | Vessel growth |
| TGF-•, •HGF | EGF-R, c-Met | endostatin | ||
| PDGF-BB | PDGF-R• • | Recruits pricytes | Stabilization of nascent vessels | |
| Plasminogen | TIMPs | Remodel matrix, | ECM remodelling and | |
| activators, MMPs | PAI-1 | release and activate growth factors | EC migration | |
| TGF-• • | Endoglin | Stimulate extracellular matrix production | Vessel growth |
Based on this knowledge, Xalcedo et al29 designed a study in order to test the hypothesis that the evaluation of angiogenesis markers might provide additional, valuable information on the evolution of liver lesion in CHC after antiviral therapy. They included 36 naïve HCV-infected patients and 15 healthy individuals. HCV-infected patients presented a «proangiogenic» profile of angiogenesis soluble markers (VEGF and Ang-2), different from that in healthy controls, while sTie-2 serum levels were similars in both groups. Their results showed a significant correlation between the grade of inflammation in biopsies and serum levels of VEGF, which suggest that inflammation and angiogenesis processes are strongly associated in those patients.
Interestingly, treatment of CHC patients with pegylated interferon plus ribavirin significantly reduced VEGF and Ang-2 levels with respect to pretreatment values. Mean sTie-2 levels were significantly increased after therapy. Taken together, these data indicate that antiviral combination therapy determines a shift toward an «antiangiogenic» profile of serum markers in CHC patients. The therapy-induced decrease in VEGF levels was significantly correlated with that in ALT and alkaline phospatase serum levels in responder patients, but not in nonresponders. A similar correlation was found between the decreases in the serum levels of Ang-2 and ALT. These significant relationships suggest some degree of association between angiogenesis and the extent of liver damage (Figure 1).
Their novel findings have important implications.29Firstly, the demonstration of a close relationship between variations in serum levels of angiogenesis markers and response to therapy in CHC patients conceptually supports the clinical role of angiogenesis in chronic inflammatory liver disease. Secondly, the determination of VEGF, Ang-2, and sTie-2 in parallel to the viral load and fibrosis markers may provide complementary information to asses response to treatment and disease progression, which could be helpful to define the optimal follow-up therapeutic strategy for each patient. Further prospective studies with larger cohorts of patients are required to investigate whether these markers may have prognostic value. Additionally, based on these findings, it may be worth investigating in detail the antiangiogenic effects of pegylated interferons and, hypothetically, to consider whether antiangiogenic drugs targeting VEGF or Ang-2 could be of additional value for CHC patients with advanced liver disease and those presently considered as «difficult to treat».
Angiogenesis in autoimmune liver diseaseControversy exists regarding intrahepatic vasculature in autoimmune liver disease. Some authors have reported a tendency to vasopenia and decreased peribiliary capillary plexus in the livers of primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), and autoimmune hepatitis (AIH) patients.30,31 This has been attributed to destruction of vascular estructures by autoimmune mechanisms, similar to the process undergone by bile ducts in PBC and PSC. However, evidence of angiogenesis has been observed in PBC and AIH. Angiogenesis may occur at a later stage, in response to the hypoxia caused by depletion of existing vessels, although the precise kinetics of the process have not been investigated.
PBC is a chronic inflammatory liver disease of multifactorial etiopathogenesis, characterized by the presence of an intrahepatic mononuclear cell infiltrate, as well as circulating autoantibodies.32,33 It has been suggested that immunological mechanisms involving T-lymphocytemediated lysis are important in the characteristic bileduct and hepatocellular damage occuring in PBC.34-36Medina et al37 evaluated the phenotype of cellular infiltrates, and the expression of lymphocyte activation, antigen recognition and cell-adhesion molecules in the livers of PBC patients; and they investigated and characterized at the molecular level the pattern of reactivity of proangiogenic factors involved in the formation of neovessels in PBC liver samples. The ocurrence of angiogenesis in the livers of PBC patients was a novel finding of this study. CD31 and VE-cadherin positive EC assemble to form new vascular structures, mainly in portal and periportal areas, in association with inflammatory infiltrates and fibrosis. They observated an enhanced expression of VEGF, Ang-1, Ang-2, their receptor Tie-2 and endoglin, which suggests their involvement in EC proliferation and nascent vessels stabilization.
AIH is a chronic progressive liver disease characterized by serological changes (hypergammaglobulinemia, autoantibodies) and interface hepatitis on histological analysis.1 Tubular structures reflecting formation of new vasculature are observed in inflamed portal tracts of AIH patients.38 As in other inflammatory liver diseases, an upregulation of inducible NO synthase leading to an enhanced production of NO occurs in AIH, and this may participate in angiogenic processes.39 However, information about the mechanisms of angiogenesis in AIH is still very limited.
Angiogenesis in hepatocellular carcinomaZhang et al40 evaluated the expression of angiogenic factors in hepatocellular carcinoma (HCC) compared with the nontumour liver tissue. They found a high expression of Ang-2 and low expression Ang-1 in HCC samples in comparison with nontumorous tissue, indicating that they play a key role in the carcinogenesis and progresion of HCC via angiogenesis. Pappeti and Herman described that tumorous angiogenesis is very different from the physiological process.41 During this process, vascular quiescence and stabilization are mediated by Ang-1, Ang-2 and Tie-2 system. Therefore, the pathologic state of imbalanced Ang-2/Ang-1 ratio in the presence of VEGF plays a critical role in the transformation of non-cancerous liver to liver cancer by initiating early neovascularization. Vajkoczy et al42 reported that tumors in their very early stage are initiated by host vessels via VEGF, VEGF receptor-2 and Ang-2.
In conclusion, expression of Ang-2 against Ang-1 through the Tie2 receptor in the presence of VEGF plays a critical role in initiating early neovascularization and induces transformation of noncancerous liver to HCC.
ConclusionsIn recent years, it has become increasingly evident that pathological neoangiogenesis processes occur in chronic inflammatory and fibrotic liver diseases (and probably will be discovered in other nontumoral hepatic alterations). The challenge for the upcoming years is the characterization of the molecular basis and pathways of angiogenic disorders in an integrated manner. The angiogenesis in chronic inflammatory liver diseases seems to have a prognostic value in the evaluation of disease progression. A better understanding of the process may also lead to the design of efficient and safe antiangiogenic therapies using appropriate combinations of inhibitors of angiogenesis. Antiangiogenic therapy is an effective novel treatment for HCC, which is of great clinical significance.43



