



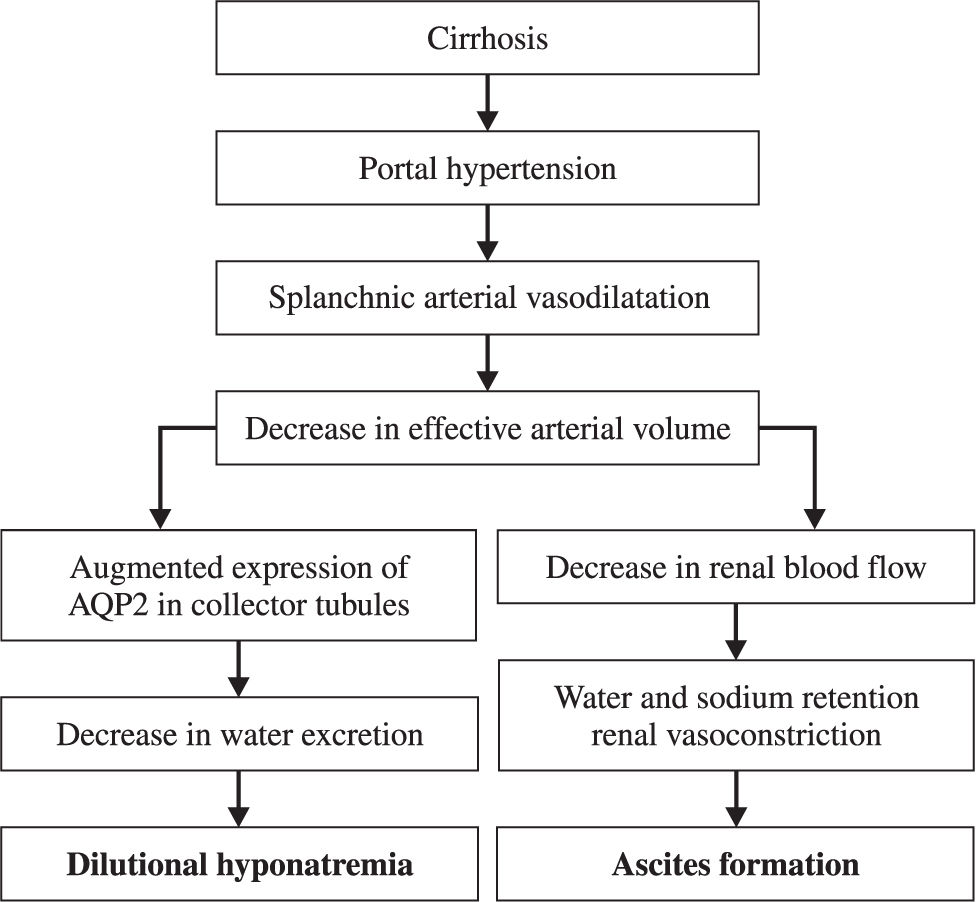

Ascites, the most common complication of cirrhosis, is associated with a poor quality of life, an increased risk of infection, and renal failure. Twenty percent of cirrhotic patients have ascites at the time of diagnosis, while 30% and 50% will develop ascites by 5 and 10 years, respectively. There are several factors that contribute to ascites formation in cirrhotic patients, these include splanchnic vasodilatation, arterial hypotension, high cardiac output, and decreased vascular resistance. These factors lead to ineffective intravascular volume (hyperdynamic state), impairment of renal function, and subsequent water and sodium retention, all of which lead to dilutional hyponatremia (serum sodium <130 mEq/L), one of the most important prognostic factors in these patients. In conclusion, the therapeutic objective is to improve sodium balance and circulatory function through non-pharmacological measures, such as dietary sodium and water restriction as well as bed rest. Spironolactone (100-400 mg/day) is the initial drug of choice, while loop diuretics (like furosemide, 40-60 mg/day) are frequently used as adjuvants. Recently, agents that interfere with the renal effects of vasopressin by inhibiting water reabsorption in collecting ducts and producing free water diuresis have been used. These agents are called aquaretics and can be useful in the treatment of ascites unresponsive to conventional therapy.
Abbreviations
RAAS: renin-angiotensin-aldosterone system
SNS: sympathetic nervous system
ADH: antidiuretic hormone
ANP: atrial natriuretic peptide
CGRP: calcitonin gene related peptide
AQP: aquaporine
cAMP: cyclic adenosine monophosphate
EABV: effective arterial blood volume
IntroductionCirrhosis is an expanding problem and has multiple etiologies. Most of the morbidity and mortality of chronic liver diseases is due to its progression to cirrhosis and complications of cirrhosis. Ascites is the most common major complication of cirrhosis; other complications include hepatic encephalopathy and variceal hemorrhage. Ascites is associated with poor quality of life, increased risk of infection, and renal failure. Twenty percent of patients with cirrhosis have ascites at the time of the diagnosis. While 30% and 50% of patients with compensated cirrhosis will develop ascites in 5 and 10 years of followup, respectively. Ascites, which is a sign of poor prognosis, characteristically develops during late stages of the disease. Patients with cirrhosis and ascites have 50% mortality at two years.1,2
Dilutional hyponatremia is defined as serum sodium < 130 mEq/l secondary to excess total body water in patients without sodium deficit or diuretic therapy. The incidence of dilutional hyponatremia in cirrhotic patients being treated for an episode of ascites is 35%, and it is one of the most important prognostic factors in these patients.3
Pathophysiology of ascites in cirrhosisThere are several factors involved in the development of ascites in cirrhotic patients. The principal pathophysiological factors are severe sinusoidal portal hypertension and liver failure.
These factors lead to circulatory dysfunction, characterized by splanchnic arterial vasodilation, arterial hypotension, increased cardiac output, and decreased vascular resistance. All these factors lead to ineffective intravascular volume and impairment of renal function that cause subsequent water and sodium retention.4
The most important theories of ascites formation in cirrhotic patients are described in the subsequent paragraphs.
Underfilling hypothesisThis theory suggests that transudation of water and sodium into the abdominal cavity, due to hepatic venous obstruction and portal hypertension, produces hypovolemia and secondary retention of sodium and water.
The forces that regulate the movement of fluid in the extracellular compartment are called Starling forces. These forces include the hydrostatic pressure of the vascular system, the oncotic pressure of interstitial fluid, the oncotic pressure of plasma proteins, and the hydrostatic pressure of interstitial fluid (tissue tension). The first two forces facilitate the movement of fluid from the vascular space to the extravascular space and the last two favor the movement of fluid towards the intravascular space.
Portal hypertension (increased hydrostatic pressure of the vascular system) and hypoalbuminemia (decreased oncotic pressure of plasma) disrupt the Starling equilibrium in the splanchnic microcirculation. This imbalance could result in increased production of splanchnic lymph.
When portal hypertension is mild to moderate, increased lymphatic drainage through the thoracic duct compensates excess fluid formation in the abdominal cavity. However, when portal hypertension is severe, lymph overflow causes the movement of fluid from the interstitial space of splanchnic organs to the abdominal cavity, which secondarily produces impairment of circulatory function and ascites. Increased plasma volume and cardiac output as well as decreased peripheral vascular resistance in cirrhotic patients are arguments in favor of this theory5,6
Overflow hypothesisThis theory, proposed by Liberman et al, is based on the fact that cirrhotic patients with ascites have increased total blood volume. The overflow hypothesis suggests that ascites formation is not related to decreased intravascular volume and rather is a secondary phenomenon due to «primary» retention of sodium and water. It proposes that the cause of this «primary» retention is a «hepatorenal reflex» that predominates over the normal volume regulation mechanism.7
Peripheral arterial vasodilation hypothesisThis theory includes characteristics of both theories and considers that splanchnic arteriolar vasodilation secondary to portal hypertension is the initial event. Cardiac preload decreases with a subsequent increase in cardiac output and activation of the systems that produce vasoconstriction (renin-angiotensin-aldosterone system, sympathetic nervous system, and vasopressin).
During the initial stages of cirrhosis, when there is no ascites, circulatory homeostasis is maintained by hyperdynamic circulation (high plasma volume and high cardiac output). With disease progression, compensatory arterial splanchnic vasodilation increases, yet it is insufficient to maintain circulatory homeostasis and arterial hypotension develops. Afterwards, several mechanisms are activated to try to maintain normal arterial pressure. The principal blood pressure regulatory mechanisms are the rennin-angiotensin-aldosterone system (RAAS), the sympathetic nervous system (SNS), and the antidiuretic hormone (ADH).5,6 The mechanism by which portal hypertension is associated to arterial splanchnic vasodilatation and the resistance that splanchnic arterioles have to vasoconstrictors are not totally understood. Some studies suggest that it could be related to increased levels of circulating vasodilators (glucagon, prostaglandins, atrial natriuretic peptide (ANP), calcitonin gene-related peptide (CGRP)).
Nevertheless, other authors argue that excess synthesis or release of local vasodilators (nitric oxide) into the vascular splanchnic compartment is a predominant factor. No explanation is known for the increased local activity of vasodilators in portal hypertension.4,8-10
Therefore, in compensated cirrhosis (without ascites), peripheral arterial vasodilatation (underfilling) is the first event leading to decreased intravascular blood volume. This vasodilation, related to the «underfilling» of arterial circulation, is associated to compensatory mechanisms previously mentioned.
In order to increase intravascular volume, cardiac output increases and the kidney retains water and sodium. In this stage of cirrhosis there is increased total blood volume, cardiac output, and peripheral vasodilation. These «compensated cirrhotics» (without diuretic therapy) do not form ascites if a low dietary ingestion of salt is maintained. At this stage, the plasma levels of renin, aldosterone, vasopressin, or norepinephrine are not elevated.
This hypothesis proposes a transient elevation of the aforementioned hormones during compensatory retention of sodium and water by the kidney. This elevation leads to volume expansion and return to normal hormone levels. On the other hand, decompensated cirrhosis is defined by ascites formation. It represents an advanced stage in the arterial vasodilation hypothesis. At this stage, increased blood volume secondary to transitory renal retention of water and sodium is not enough to maintain circulatory homeostasis. In addition, decreased plasma oncotic pressure could be an additional factor contributing to decreased effective arterial blood volume. Therefore, when arteriolar baroreceptors detect diminished filling of the arterial tree, vasopressin is secreted and the renin-angiotensin-aldosterone and sympathetic systems are activated. These hormones are not elevated in approximately 25% of patients with decompensated cirrhosis, probably due to less severe peripheral vasodilation. Twenty-five percent of patients with decompensated cirrhosis who have high plasma levels of vasoconstrictors have normal renal perfusion. The later is attributed to compensatory intrarenal vasodilator systems, mainly prostaglandins and the kinin-kallikrein system. Therefore, prostaglandin synthetase inhibitors are contraindicated in these patients.10-12
Water and sodium retention mechanismsDeterioration of the capacity to excrete sodium is the first renal disturbance in cirrhotic patients. It is mainly due to increased tubular reabsorption of sodium. In advanced stages of the disease, there are considerable decreases in renal blood flow and glomerular filtration (GF). They are due to vasoconstriction of renal arteries by way of intrinsic mechanisms and antinatriuretic systems, especially RAAS and the sympathetic nervous system. Therefore, patients with cirrhosis and ascites have hyperaldosteronism and sympathetic nervous system hyperactivity.5,13
Hyperaldosteronism stimulates the reabsorption of sodium in the distal nephron, while the sympathetic nervous system increases the reabsorption in the proximal convoluted tubule, the Henle loop, and the distal nephron. These are the two most important factors favoring water and sodium retention in patients with cirrhosis. In addition, circulating levels of ANP are augmented. This concept suggests that the effects of anti-natriuretic systems predominate over those of endogenous natriuretic hormones.4,14
These mechanisms also contribute to reduction of free water clearance by the kidney. The principal clinical consequence of this reduction is the impairment of dietary water excretion. This contributes to ascites formation and the dilution of corporal fluids, producing hypotonic hyponatremia. Dilutional hyponatremia (Na < 130 mEq/L) is a late event in the course of decompensated cirrhosis and indicates a low probability of survival.3,15
Antidiuretic hormone activationADH or vasopressin is a hormone that is synthesized in the supraoptic and paraventricular nuclei of the hypothalamus and is transported through the axons of these nuclei to the posterior pituitary gland. The principal factors that contribute to the release of ADH are plasma osmolality and the circulatory volume.4
The neurons known as osmoreceptors, concentrated in the hypothalamus, are sensitive to changes in plasma osmolality. The molecular mechanisms for this sensitivity are not clear, however, some studies have demonstrated that these cells have mechanosensitive ion channels that quickly respond to changes in volume. Aquaporine-4, a member of the family of water channel proteins is highly expressed in the plasma membrane of these osmoreceptors and can mediate rapid changes in cellular volume in response to local disruption of the extracellular osmolality. In addition to hypothalamic osmoreceptors, secretion of vasopressin is also controlled by non-osmotic stimuli through the autonomous nervous system. Baroreceptors, the primary receptors of this mechanism, communicate with the hypothalamus through parasympathetic nerves and are located in the atrium, ventricle, aortic arch, and carotid sinus.3,4
ADH increases the permeability of the distal convoluted tubule and collecting duct to increase the reabsorption of water. Another transcendental function is vasoconstriction (which is responsible for the name vasopressin). The first biological step is the binding of vasopressin with V2 receptors on the basolateral membrane of renal collecting ducts.
Experimental studies indicate that ADH has a fundamental role in ascites formation in cirrhotic patients. Plasma levels of ADH are elevated in late stages of the disease and correlate with the renal capacity to excrete free water.
As mentioned previously, massive release of ADH in cirrhotic patients with ascites occurs as a mechanism to maintain arterial pressure. The principal factor for ADH secretion is non-osmotic stimuli, in other words, hemodynamic (arterial hypotension).16 This is important in order to devise specific pharmacological therapies directed at the different mechanisms through which ascites is formed.
Evolution of dilutional hyponatremiaSodium retention is the most frequent electrolyte disorder associated to cirrhosis and it leads to the development of edema and ascites. Another common problem is water retention, which also leads to hyponatremia (Figure 1). Hyponatremia in patients with cirrhosis is dilutional and therefore fluid restriction is the cornerstone of treatment.3,17
Therapeutic recommendations for ascites
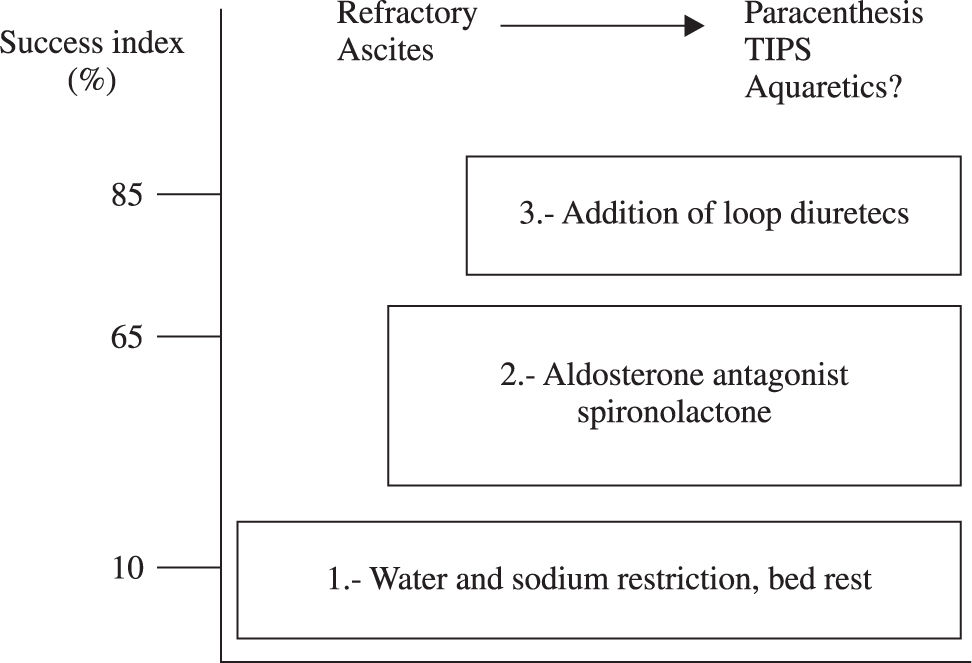
There are two types of ascites in the cirrhotic patient, uncomplicated ascites and refractory ascites2,18-20(Figure 2).

- 1.
Uncomplicated ascites: Ascites that is not infected and is not associated to hepatorenal syndrome. It is divided into 3 groups:
- a.
Grade 1 (mild): Only detected on ultrasound examination.
- b.
Grade 2 (moderate): Manifested by symmetric distention of the abdomen.
- c.
Grade 3 (severe): Gross ascites with marked abdominal distention.
- a.
- 2.
Refractory ascites: It was defined in 1996 by the International Ascites Club2,18-20 as ascites that cannot be mobilized or the early recurrence of which (i.e., after therapeutic paracentesis) cannot be satisfactorily prevented by medical therapy. It occurs in 5% to10% of all cases with ascites. Two subgroups were identified:
- a.
Diuretic-resistant ascites: ascites that cannot be mobilized or the early recurrence of which cannot be prevented because of lack of response to dietary sodium restriction and intensive diuretic treatment.
- b.
Diuretic-intractable ascites: ascites that cannot be mobilized or the early recurrence of which cannot be prevented because of the development of diuretic-induced complications that preclude the use of an effective diuretic dosage.
- a.
The objective of treatment is to improve sodium balance and circulatory function until orthotopic hepatic transplantation is available. Non-pharmacological measures, such as bed rest, dietary sodium, and water restriction are recommended (in the case of dilutional hyponatremia).20 The administration of diuretics (spironolactone 100-400 mg/day in combination with the loop diuretic furosemide 40-160 mg/day)21 and paracentesis can also be of use18,22,23, Table I).
Treatment of ascites and mechanism of action.
| Management | Mechanism of action | Recommendation |
|---|---|---|
| Bed rest | Standing position activates systems that favor sodium retention | Doubtful utility |
| Water and sodium restriction | Negative balance between ingestion and renal excretion | Ascites: 5.2 g of salt per day Dilutional hyponatremia: hydric restriction |
| Aldactone | Blocks the renal retention of sodium in distal tubules and collector tubules of the nephron induced by the secondary hyperaldosteronism. | 100-400 mg/day Only in combination with furosemide |
| Amiloride | Similar to aldactone, with less adverse reactions (gynecomastia) | 20-60 mg/day |
| Furosemide | Loop diuretic with renal excretion of sodium. | 40-120 mg/day used as an adjuvant on the therapy with spironolactone |
| Paracenthesis | Removes great quantities of ascites liquid | In extractions of > 5L it is recommended the reposition with albumin with a dose of 8g per liter of ascites fluid removed. |
Note: The weight of the patient is daily measure to assess mobilization of ascites fluid. The loss of weight index must be 500 g/day in the absence of peripheral edema and 1 Kg/day when edema is present. The response to treatment is assessed with the excretion of urinary sodium. An excretion of > 90 mmol/day in urine in a patient that does not loses weight suggests that he does not follow the diet in an adequate way; on the other hand, an urinary excretion of < 90 mmol/day in a subject with increase in weight and persistent ascites traduces diuretic treatment resistance.
Recently, aquaretic agents have been investigated for the treatment of ascites unresponsive to conventional treatment. These agents will be described with more detail in the subsequent text.
Role of aquaporinsAquaporins are membrane proteins that form in channels and facilitate the movement of water through lipidic membranes by means of osmotic or hydraulic gradients. These molecules are expressed by multiple tissues and are essential for corporal water homeostasis. The majority of these molecules are located in the kidney.24,25
In 1992 Agree et al. discovered the first water channel (CHIP28 protein), which was later named aquaporin-1 (AQP1). With this discovery, research in water homeostasis and its movement through lipidic membranes commenced.26 Up to now, 11 types of aquaporins have been described (AQP 0 to 10). They are divided in two main groups, the first of which are exclusively permeable to water (AQP0, AQP1, AQP2, AQP4, AQP5, AQP6 and AQP8) and the second of which are also permeable to glycerol (AQP3, AQP7, AQP9 and AQP10).
Aquaporin water channel proteins are simple polypeptide chains of approximately 28Kda that have 6 transmembrane domains and whose amino and carboxyl termini portions are intracytoplasmic. Aquaporins have a three-dimensional structure that forms tetramers in the plasma membrane, except aquaporin-4, which has an orthogonal structure. The AQP1 pore has a diameter of 3Å in its narrowest portion, while a water molecule measures 2.8Å. The calculated velocity with which water molecules pass through aquaporins is 3x109 molecules per monomer per second.25 Aquaporin-2 is regulated by vasopressin and the antagonism of this aquaporin is of special interest in patients with edematous states like cardiac failure and hepatic cirrhosis. Therefore it will be emphasized in this review.
Aquaporin 2 (AQP2)Type 2 aquaporins (AQP2) are found in the apical membrane, in subapical intracytoplasmic vesicles, and in the basolateral plasma membrane of the renal collecting ducts. The amino and carboxyl termini of this aquaporin are involved in the apical arrangement of the cell, however, the exact mechanism of action has not been clarified yet. Experimental models in rats with cirrhosis have demonstrated altered water excretion dependent on aquaporine-2 expression in the renal collecting ducts.27,28
Short-term and long-term pathways regulate the expression of AQP2.
Short-term regulationWhen vasopressin binds to the V2 receptor on the basolateral membrane of the collecting ducts, adenyl cyclase is activated by means of a G protein (GTP-binding protein), which produces an increase in cAMP. cAMP then binds to the regulatory unit of the kinase protein A (KPA) and allows the dissociation of the regulatory and catalytic units of this kinase protein. The catalytic subunit produces phosphorylation of AQP-2 at the serine residue 256 from the carboxyl-terminus. Intracytoplasmic vesicles of phosphorylated AQP-2 are then translocated to the apical plasma membrane where AQP2 increases the collecting duct’s permeability to water. At least 3 of the 4 monomers of the AQP2 must be phosphorylated in order to be inserted into the apical membrane. The transportation and insertion of the AQP2 in the apical plasma membrane is mediated by microtubules and actin. When plasma vasopressin concentrations decrease or the V2 receptor is blocked, the AQP2 suffers endocytosis, which causes the tubular epithelium to decrease its permeability to water.
Long-term regulationIn addition to the short-term regulation, long-term regulation of AQP2 expression is controlled by the state of hydration. In other words, water restriction increases AQP2 expression in the cell and produces vasopressin release. And as previously described, vasopressin increases AQP2 mRNA transcription.29
Aquaretic agentsAquaretic agents inhibit water reabsorption and produce solute-free diuresis in the collecting ducts by interfering with the renal effects of vasopressin.
During the past few decades, numerous drugs have shown the ability to produce free water excretion without affecting electrolyte balance. However, the majority of these drugs has not proven useful and has been associated with important adverse effects. The most important aquaretic agents are demeclocycline, urea, oral prostaglandins, and selective of V2 receptor antagonists.30
Vasopressin receptor antagonists are the most important aquaretic agents that have been proven useful. The k-opioid receptor agonists have also been shown useful, although to a lesser degree. The utility of these aquaretic agents on body water homeostasis has been demonstrated in cirrhosis. However, its clinical utility in patients with cirrhosis, ascites, and hyponatremia remains to be defined in a controlled clinical trial.
OpioidsAs shown over twenty years ago, opioid peptides have an important effect on water excretion in addition to their known analgesic and psychotropic effects. Activation of mu receptors stimulates the secretion of vasopressin, which promotes antidiuresis. Kappa-opioid agonists increase urinary volumes and reduce urine osmolarity without changing electrolyte excretion (hypotonic polyuria). They act through negative regulation of the antidiuretic hormone in the pituitary.
Niravoline (RU51599) is a K-opioid receptor agonist that induces an aquaretic response in cirrhotic patients. It is well tolerated at moderate doses (0.5-1 mg), however, reversible personality disorders and mild confusion can occur at higher doses (1.5-2 mg). Intravenous administration limits its clinical use.31
Vasopressin receptor antagonistsThese agents inhibit water reabsorption in the collecting ducts by selective antagonism of renal vasopressin V1 and V2 receptors.
Non-selective V1 and V2 receptors antagonists.Conivaptan (YMO87) was the first non-peptide antagonist of V1 and V2 receptors. It has been used in clinical studies for the management of heart failure with promising hemodynamic effects. It reduced pulmonary capillary pressure and increased diuresis without causing adverse effects such as arterial hypotension or tachycardia. It probably has less clinical benefit in cirrhosis because of its effect on V1 receptors, although it has not been evaluated in clinical studies.32
Selective V1 receptor antagonists.The initial role of these antagonists was proposed to be the management of hypertension and congestive heart failure. Nevertheless, SR49059 did not prove clinical utility as monotherapy in clinical studies. Therefore the role of these antagonists has not been thoroughly defined.33
Selective V2 receptor antagonistsThese antagonists can be useful in the management of advanced cirrhosis with ascites and hyponatremia. The first selective antagonist of V2 receptors was a peptide and was a modification of the desmopressin molecule (a selective V2 agonist). It was useful in animal models, however its usefulness in humans was not proven because of its agonistic effect on V2 receptors and its short half-life. Several oral, non-peptide, selective antagonists of V2 receptors have been subsequently developed and have proven their clinical utility. They include OPC-31260, VPA-985, SR121463, OPC-41061, YM-087, and Talvaptan.34
VPA-985 is a non-peptide, highly selective, competitive antagonist of the V2 vasopressin receptor. It has been demonstrated that this compound promotes renal excretion of water without affecting electrolyte excretion and therefore improves plasma sodium concentration in a dose-dependent manner.35 A clinical study established that VPA-985 was clinically well tolerated, reaching its maximum plasma concentration in one hour. No impairment in renal function was observed and hepatic encephalopathy was reported only in a small percentage of the patients.36
Only 50% of the cases with cirrhosis and hyponatremia respond to aquaretic therapy. Currently, factors that can modify the response rate remain unknown.37
Because of interindividual variability in the effects of these drugs, low-dose therapy is initially recommended with progressive dose escalation. Plasma sodium should be monitored to avoid an excessive correction that could lead to central pontine myelinolysis.38 The duration of the aquaretic effect during chronic administration should be confirmed by prospective studies in cirrhotic patients.
Clinical studies have demonstrated a diuretic effect with the combination of vasopressin and furosemide or hydrochlorothiazide. This combination increases treatment efficacy and could prevent diuretic-induced hyponatremia that occurs in 25% of the patients.39,40
Besides their aquaretic effect, V2 receptor antagonists also increase plasma vasopressin concentration, which could cause secondary vasoconstriction through activation of V1 receptors. This could be an additional benefit for patients with cirrhosis in whom vasoconstriction could lower portal pressure and prevent variceal hemorrhage, nevertheless this should be confirmed with prospective studies.34
Recently, a randomized, placebo-controlled study found that 30% and 22% of cirrhotic patients with hyponatremia (serum Na < 130 mEq/L) who received Talvaptan had normalized their plasma sodium (> 135 mE/ L) by day 4 and 30, respectively. However, up to 22% presented severe adverse effects such as hypotension, encephalopathy, gastrointestinal bleeding, worsening of heart failure, and death. These adverse reactions could limit its use in clinical practice.41
Potential clinical uses of aquaretics in cirrhotic patients- 1.
Diuretic-refractory ascites and edema
- 2.
Hyponatremia (serum Na < 130 mEq/L)
- 3.
As adjuncts to standard diuretics
Ascites and dilutional hyponatremia are frequent complications in cirrhotic patients that are associated with a decrement in quality of life and renal function. Nowadays loop diuretics (furosemide) and aldosterone antagonists (spironolactone) are the treatment of choice, yet are not exempt of adverse effects. Some patients do not respond to conventional treatment, therefore new therapeutic options are necessary. Aquaretic agents inhibit water reabsorption in the collecting ducts, produce water diuresis free of electrolytes, and are potentially useful in the management of cirrhotic patients with refractory ascites and dilutional hyponatremia. Future studies with these agents are necessary to confirm their clinical benefit.
GlossaryDilutional hyponatremia: serum Na < 130 mEq/L in the absence of diuretic treatment.
Not complicated ascites: not infected ascites and not related to the development of hepatorenal syndrome. It is divided in 3 grades:
- •
Grade 1 (mild): only detected on ultrasonography
- •
Grade 2 (moderate): manifested by symmetrical abdominal distention
- •
Grade3 (severe): marked abdominal distention
Refractory ascites: ascites that cannot be mobilized or with early recurrence under optimal treatment. Two subgroups are identified in this type of patients:
- 1)
Diuretic resistant ascites: is the ascites that cannot be eliminated or which recurrence cannot be avoided due to a lack of response to salt restriction and maximal doses of diuretics.
- 2)
Ascites that cannot be treated with diuretics: is the ascites that cannot be eliminated or which recurrence cannot be avoided due to the development of complications induced by diuretics that impede the use of these drugs.
Aquaporines: channel-shaped membrane proteins that facilitate the movement of water through lipidic membranes by the means of osmotic or hydraulic gradients.
Aquaretics: agents that interfere with the renal effects of vasopressin inhibiting water reabsorption in the collector tubules so they produce water diuresis free of solutes.
Effective arterial blood volume (EABV): the part from the total arterial volume that is capable of stimulating the volume receptors and stimulate intrarenal mechanisms of water and sodium retention.



