




Liver regeneration plays a valuable significance for hepatectomies, and is mainly attributed to hepatocyte proliferation. MicroRNA-125a-3p was reported to be highly associated with liver regeneration process. We studied the underlying mechanism of the functional role of miR-125a-3p in liver regeneration.
Materials and methodsThe miR-125a-3p mimics and inhibitor vector were constructed and transfected into primary human liver HL-7702 cells, the transfected cell viability was detected using cell counting kit-8 (CCK-8). Cell cycle distribution was analyzed by flow cytometry. With Targetscan and OUGene prediction, the potential targets of miR-125 were verified by real-time quantitative PCR (qPCR) and luciferase reporter assays in turn. The overexpression vector of proline-rich acidic protein 1 (PRAP1) was constructed and co-transfected with miR-125a-3p mimics into HL-7702 cells, detecting the changes of proliferative capacity and cell cycle distribution. Western blot and qPCR performed to analyze gene expressions.
ResultsOverexpressed miR-125a-3p notably increased the hepatocyte viability at 48h, and decreased the number of G1 phase cells (p<0.05). However, miR-125a-3p inhibition suppressed the development of hepatocytes. PRAP1 was the target of miR-125a-3p. After co-transfection with PRAP1 vector, hepatocyte viability was decrease and the G1 phase cell number was increased (p<0.05). More importantly, overexpressed PRAP1 notably decreased the mRNA and protein levels of cyclin D1, cyclin-dependent kinase 2 (CDK2) and cell division cycle 25A (CDC25A).
ConclusionThe elevated miR-125a-3p positively correlated with hepatocyte viability and cell cycle progression due to the modulation of PRAP1, and miR-125a-3p may contribute to improving liver regeneration.
The liver, one of the most vital organs in the body, occupies a central place in metabolic homeostasis [1,2]. Hepatocytes are quiescent in the normal adult liver, however, the hepatic tissue injury or other stimulation can induce hepatocytes to re-enter cell cycle and proliferation to recover the liver function [3]. Currently, for several liver diseases, such as liver neoplasms, liver cirrhosis, and cholangiocarcinoma, hepatectomy has become a common treatment [4]. After hepatectomy, the liver regeneration process was activated to help residual liver restore the proper architecture and function. It has been reported that the loss of hepatocytes by hepatectomy may induce liver injury and ultimately lead to treatment failure in patients with liver disease [5]. When the capacity of liver regeneration was not sufficient to restore the complete structure and function of the liver from the residual liver volume, the patients will die for the liver failure [6]. As the main compensatory method after hepatectomy, investigating the underlying mechanism of liver regeneration is extremely necessary for more effectively activating liver regeneration and improving the treatment of liver failure.
Liver regeneration is a complex and progressive process and it mainly relies on the hepatocyte proliferation [7]. It was documented that various types of proliferation-promoting factors were activated and involved in the regulation of hepatocyte cell cycle progression during the liver regeneration [8], for example, interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) has been proved to participate in the initiation step of liver regeneration, and both of them could induce the G0/G1 transition of quiescent hepatocytes [9,10].
MicroRNAs (miRNAs) are a type of 20–25 nucleotide noncoding RNAs, and it played a vital role in controlling gene expressions through directly targeting to the 3′-untranslated regions (UTR) of their mRNAs. Recently, through the comparative analysis of the global miRNA expressions and proteins in the regenerating livers, several miRNAs was determined to highly associated with the priming phase of liver regeneration, among these miRNAs, the increased miR-125a level at 2h after hepatectomy indicated that miR-125a had a positive role in liver regeneration process [11]. In addition, there are some documents about the anti-cancer ability of miR-125a-3p, for example, miR-125a-3p impeded the tumor proliferation and metastasis, and decreased survival rate through blocking proto-oncogene tyrosine-protein kinase Fyn (Fyn) expression [12], and the increased miR-125a-3p could also limit the development of non-small-cell lung cancer (NSCLC) [13]. However, the study about the association between miR-125a-3p and the process of liver regeneration is still very little. In this study, we tried to identify which gene miR-125a-3p directly target to, and the molecular mechanism of miR-125a-3p acting to the liver regeneration process.
2Material and methods2.1Cell culture and transfectionPrimary human liver HL-7702 cells (Shanghai Cell Bank, Shanghai, China) was cultured with Dulbecco's modified eagle medium (DMEM, #11965118, Gibco, Grand Island, NY, USA) containing high glucose, supplemented with 10% fetal bovine serum (FBS, #10099141, Gibco), and maintained at 37°C in a humidified 5% CO2 incubator. After the cells reached 90% confluence, HL-7702 cells were trypsinized and harvested for transfection. The miR-125a-3p mimics (sense: 5′-ACAGGUGAGGUUCUUGGGAGCC-3′ and antisense: 5′-CUCCCAAGAACCUCACCUGUUU-3′), miR-125a-3p inhibitor (5′-GGCUCCCAAGAACCUCACCUGU-3′), negative control were obtained from GenePharma (Shanghai, China). Full-length proline-rich acidic protein 1 (PRAP1) overexpression vector (sense: 5′-GCAAGCTTATGAGGAGGCTCCTCCTGGTC-3′ and antisense: 5′-GCGGATCCCTACTGGGGGTGGTAGATGTGG-3′) were cloned into a pcDNA3.1 vector (Takara, Japan) and an empty pcDNA3.1 was set as negative control (NC). Then, these vectors were transfected into HL-7702 cells by Lipofectamine 2000 (Invitrogen, Carlsbad, CA) at the final concentration of 50nM, respectively. The mixture of vector and Lipofectamine 2000 transfection reagent was maintained at 37°C in 5% CO2 for 6h, and then, transfected cells were cultured with complete medium for another 24h, before the assessment of transfection efficiency.
HL-7702 cells were grouped based on the vectors, named as (A) control group: normal HL-7702 cells; (B) NC group: cells were transfected with negative control; (C) Mimics group: cells were transfected with miR-125a-3p mimics vector, (D) Inhibitor group: cells were transfected with miR-125a-3p inhibitor vector; (E) PRAP1 group: HL-7702 cells were transfected with PRAP1 overexpression vector and (F) Mimics+PRAP1 group: miR-125a-3p mimics was co-transfected with PRAP1 overexpression into HL-7702 cells.
2.2Cell viability assayFor cell viability assay, HL-7702 cells were plated onto 96-well plates (3×103cells/well). After transfection with different vectors, cells were harvested each 24h, and CCK-8 reagent was added to each well. Then, the plates were maintained at 37°C in a 5% CO2 for other 2h. Finally, optical density (OD) at 450nm was examined using the microplate reader (Multiskan FC, Thermo Scientific).
2.3Cell cycle assayCell cycle distributions of each experimental group were measured by flow cytometer through propidium iodide (PI) staining as previously described [14]. Transfected cells of each experiment groups were embedded in 6-well plates and cultured with DMEM at 37°C in 5% CO2 for 24h. After that, cells were resuspended by trypsin and washed by cold PBS, then fixed with 70% cold ethanol at 4°C overnight. After centrifugation at 1000rpm at 4°C, cell pellet was resuspended with 50μg/mL PI and 10μg/mL RNase, and maintained in dark for half an hour. OD of PI was quantified using the fluorescence-activated cell sorting by a flow cytometry (FACSCalibur, Becton Dickinson).
2.4Dual-luciferase reporter assaysTargetscan (http://www.targetscan.org/mamm_31/) and OUGene (http://www.csbio.sjtu.edu.cn/bioinf/OUGene/) were used for the prediction of potential targets of miR-125-3p, followed by verifying these predicted targets by real-time quantitative PCR (qPCR) and luciferase assay (E1910; Promega). HEK293T cells (ATCC, Manassas, VA, USA) were used for luciferase assays. In brief, 3′-UTR sequence of wild-type (WT) target genes were cloned downstream of the firefly luciferase gene in the pGL3-control vector (Promega, Madison, WI, USA), and QuickChange XL site-directed mutagenesis kit (Stratagene, Agilent Technologies, Santa Clara, CA, USA) was used to create mutant 3′-UTR plasmid mutations. HEK293T cells were plated in (5×104cells/well) a 12-well dish and incubated overnight. MiR-125a-3p mimics and WT or mutant target sequence were co-transfected into HEK293T cells via Lipofectamine 2000. After incubation at 37°C for 48h, firefly and renilla luciferase activities were determined on a Sinergy 2 luminometer (Biotek, Winooski, VT).
2.5RT-quantitative PCR (RT-qPCR)Total RNA of transfected cell samples was extracted by TRIzol (Invitrogen) according to the manufacturer's instructions, and the miRNAs were purified by mirVana miRNA Isolation kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). Firstly, cDNA was generated by miScript II RT Kit (Qiagen, Valencia, CA, USA), followed the reaction parameters: at 37°C for 60min, at 95°C for 5min, finally held at 4°C. And then, the relative levels of miRNAs were performed by miScript SYBR Green PCR kit (Qiagen). Thermocycling parameters were as follows: firstly at 95°C for 5min and 40 cycles of 95°C for 30s, 55°C for 30s and 72°C for 30s. For the relative mRNA levels, cDNA was conducted using the Prime Script RT reagent kit (Takara), followed by the reaction parameters: at 65°C for 5min, followed at 30°C for 6min and at 50°C for 60min. The relative mRNA levels were analyzed by the SYBR green detection (Takara) on an ABI PRISM® 7500 Sequence Detection system (Applied Biosystems). The 20μL volume of qPCR mixture reacted at 95°C for 2min followed by 40 cycles of 95°C for 15s and 60°C for 32s and 72°C for 30s with a final extension at 72°C for 10min. The specific primers for miRNA-125a-3p and other genes are listed in Table 1. Data were calculated by 2−ΔΔCt method [15].
Primers for RT-qPCR.
| Gene name | Primer sequences |
|---|---|
| MiR-125a-3p | Forward: 5′-ACACTCCAGCTGGGACAGGTGAGGTTCTTG-3′ |
| Reverse: 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGGCTCCCA-3′ | |
| CDC25A | Forward: 5′-CCTCCGAGTCAACAGATTCA-3′ |
| Reverse: 5′-GGGTCGATGAGCTGAAAGAT-3′ | |
| Cyclin D1 | Forward: 5′-GTCTTCCCGCTGGCCATGAACTAC-3′ |
| Reverse: 5′-GGAAGCGTGTGAGGCGGTAGTAGG-3 | |
| CDK2 | Forward: 5′-CAGTACTGCCATCCGAGAGA-3′ |
| Reverse: 5′-GAATGCCAGTGAGAGCAGAG-3′ | |
| CKAP4 | Forward: 5′-CCGTGGAATCACTCCAGAAGG-3′ |
| Reverse: 5′-AGTCCTGAGCATTTTCAAGTCC-3′ | |
| PTEN | Forward: 5′-TTTGAAGACCATAACCCACCAC-3′ |
| Reverse: 5′-ATTACACCAGTTCGTCCCTTTC-3 | |
| PARP-1 | Forward: 5′-TGATAGCAGCAAGGATCCAT-3′ |
| Reverse: 5′-TACTTCCTGATGATCTCGGCTT-3′ | |
| FBXO31 | Forward: 5′-ACTGCAAGAGTCAGGTTCCG-3′ |
| Reverse: 5′-CAAGTGTGCACTCAACTCGC-3′ | |
| P53 | Forward: 5′-TAAAAGATGTTTTGAATG-3′ |
| Reverse: 5′-ATGTGTGTGATGTTGTAGATG-3 | |
| SENP2 | Forward: 5′-GCTAAGGTTCTCGGCACCATT-3′ |
| Reverse: 5′-ATTACAAGCAGAAGACACCATG-3′ | |
| U6 | Forward: 5′-CTCGCTTCGGCAGCACA-3′ |
| Reverse: 5′-AACGCTTCACGAATTTGCGT-3′ | |
| β-Actin | Forward: 5′-CCTGGCACCCAGCACAAT-3′ |
| Reverse: 5′-GGGCCGGACTCGTCATAC-3 |
All transfected cells were resuspended with RIPA lysis buffer (Beyotime, China), and BCA Protein Assay Reagent (Pierce, Rockford, IL, USA) performed the total protein concentration of each lysate. Proteins in the lysates were electrophoretically separated by 10% sodium dodecyl sulfate-polyacrylamide gel and then transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat milk overnight at 4°C, followed by incubating overnight at 4°C with the primary antibodies. Antibodies used in this study were as followed, anti-cyclin D1 (1: 1000, #2978), anti-cell division cycle 25A (CDC25A, 1:1000, #3652) and anti-cyclin-dependent kinase 2 (CDK2, 1:1000, #2546) were obtained from Cell Signaling Technology, and anti-PRAP1 was purchased from Abcam (#ab52100, 1000, Epitomics Burlingame, CA, USA). The membranes were next incubated with horseradish peroxidase-conjugated secondary antibodies, anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, CST) and IgG H&L (HRP) (#ab205718, Abcam), at 4°C for 1h. Protein samples were visualized on an enhanced chemiluminescence (Amersham Pharmacia Biotech, Buckinghamshire, UK), and the intensities of the proteins were quantified by NIH ImageJ software (University Health Network Research, Toronto, Canada).
2.7Statistical analysisResults were presented as the mean±S.E.M. The statistical significance was performed using SPSS version 16.0 (SPSS, Inc., Chicago, IL). The difference between two independent samples was analyzed by Student's t-test. A difference was deemed statistically significant when probability value of less than 0.05.
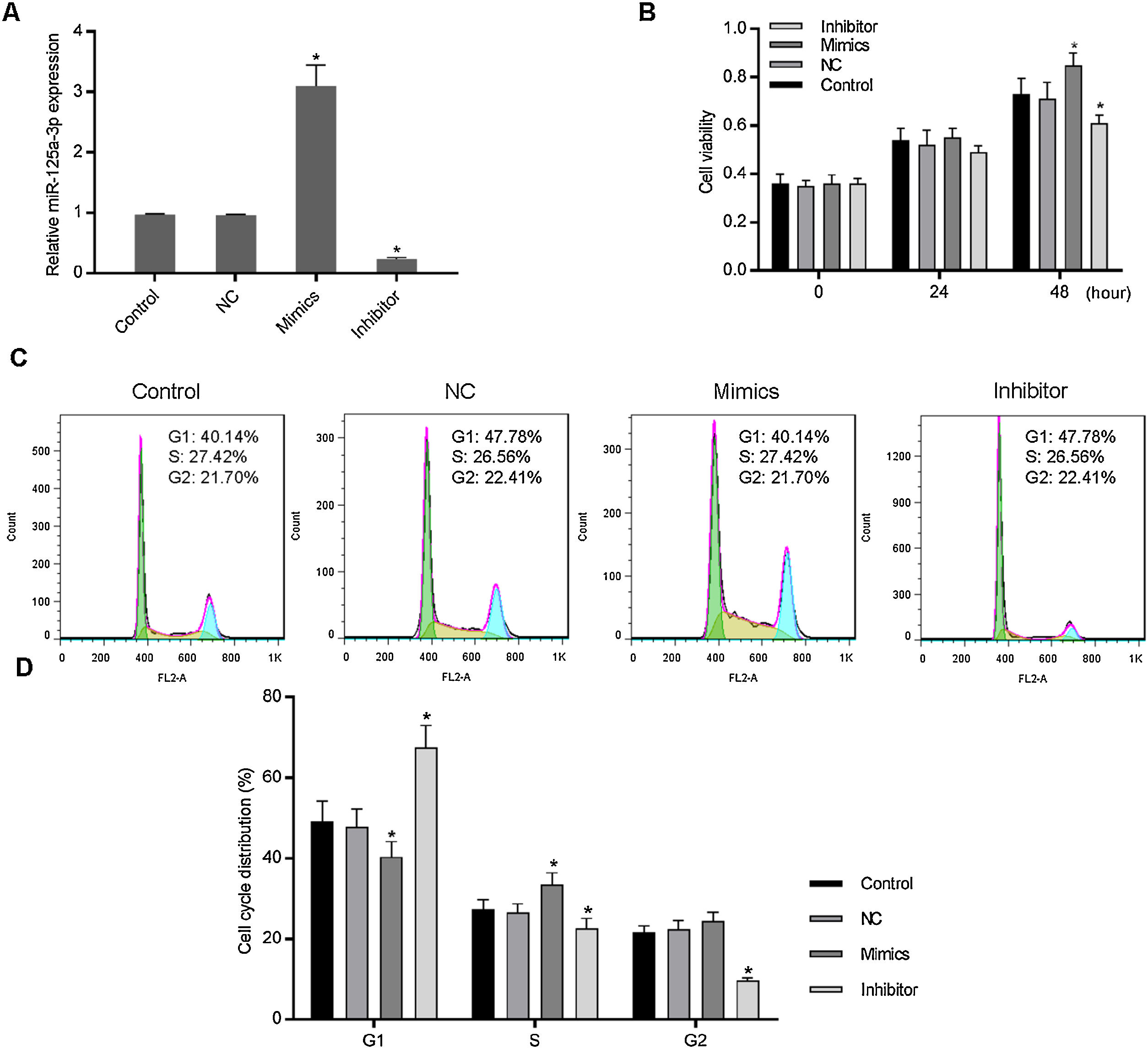
3Results3.1Increased miR-125a-3p promoted cell cycle progression of HL-7702 cellsTo investigate the functional effects of miR-125a-3p on hepatocyte proliferation during the liver regeneration, HL-7702 cells were transfected with the miR-125a-3p overexpression or inhibitor vector. The results showed in Fig. 1A indicates that both overexpression and inhibitor vectors have stably expressed in HL-7702 cells. In the meantime, we observed that overexpressed miR-125a-3p significantly elevate the HL-7702 cell viability at 48h, but miR-125a-3p inhibition down-regulated the cell viability with the incubation time increased (Fig. 1B). Additionally, the results of cell cycle distribution showed that the number of G1 phase cells was much decreased in mimics group, while the number of S phase cells was notably increased (Fig. 1C and D), however, miR-125a-3p inhibition also could effectively affect cell cycle distribution, cause G1 phase arrest and obviously decrease the number of S and G2 phase cells. Taken together, miR-125a-3p overexpression could promote the cell cycle progression of HL-7702 cells, while miR-125a-3p inhibition could lead to cell cycle G1 phase arrest.
 The transfection efficiencies of mimics and inhibitor were detected by qPCR. (B) Cell viability of transfected cells each 24h was measured by CCK-8 assay. (C and D) The effects of miR-125a-3p regulation on cell cycle distributions were analyzed by flow cytometry. Each value represents mean±SEM (n=3). U6 was considered as an internal control. *p<0.05 vs. Control group.")
Increased expression of miR-125a-3p could promote cell cycle progression in HL-7702 cells. To explore the functional role of miR-125a-3p in the process of liver regeneration, the miR-125a-3p mimics and inhibitor were constructed and transfected into HL-7702 cells. (A) The transfection efficiencies of mimics and inhibitor were detected by qPCR. (B) Cell viability of transfected cells each 24h was measured by CCK-8 assay. (C and D) The effects of miR-125a-3p regulation on cell cycle distributions were analyzed by flow cytometry. Each value represents mean±SEM (n=3). U6 was considered as an internal control. *p<0.05 vs. Control group.
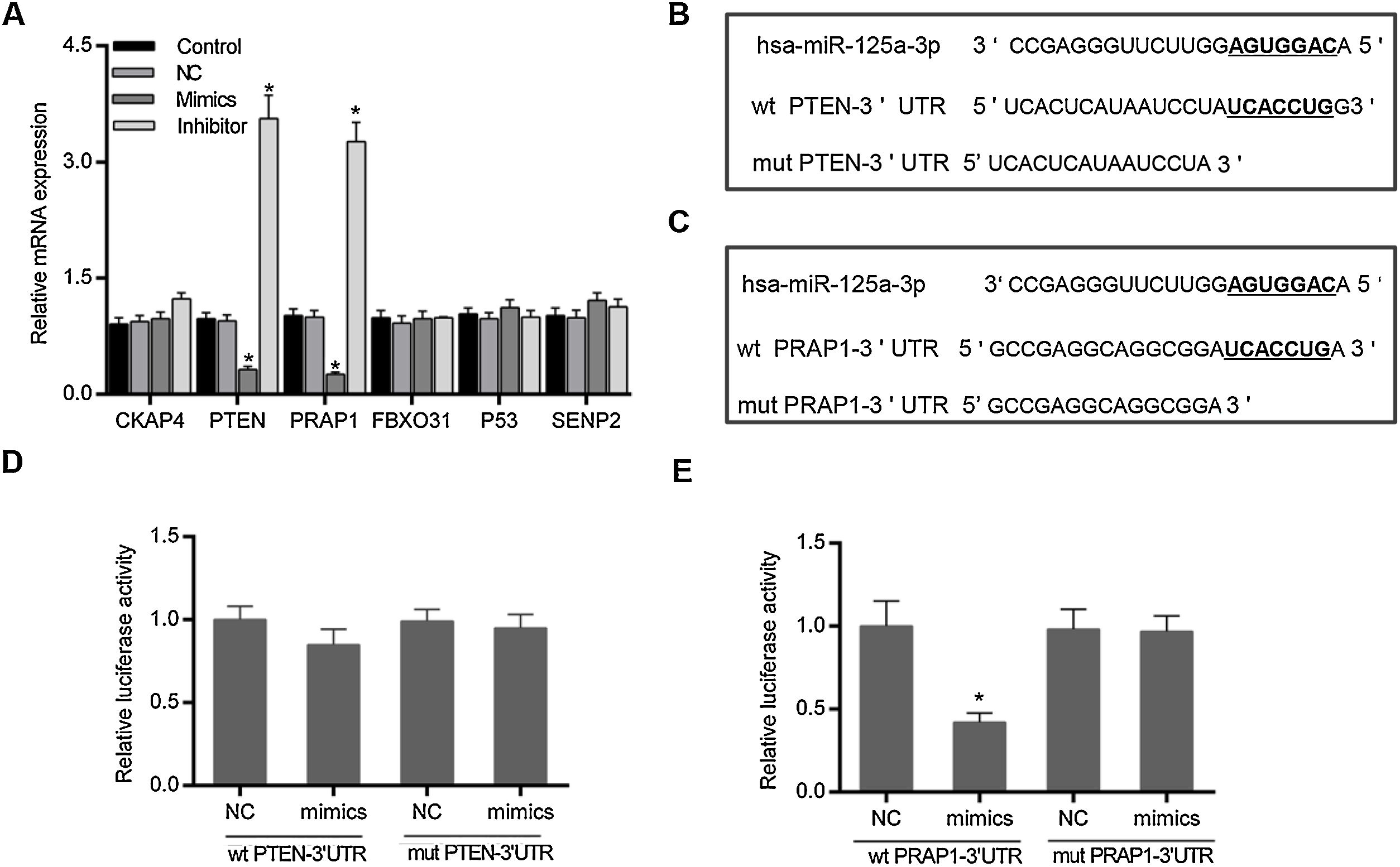
Based on the prediction of Targetscan and OUGene, six genes was supposed to be the possible targets of miR-125a-3p, including cytoskeleton-associated protein 4 (CKAP4), phosphatase and tensin homolog (PTEN), proline-rich acidic protein 1 (PRAP1), f-box only protein 31 (FBXO31), P53 and sentrin-specific protease 2 (SENP2). After the verification of qPCR, only the mRNA levels of PTEN and PRAP1 was significantly decreased in mimics group and increased in inhibitor group (p<0.05 Fig. 2A). We also observed that both the 3′-UTR sequences of wild-type PTEN and PRAP1 contained a seven-nucleotide binding site of miR-125a-3p, while these mutations of PTEN and PRAP1 could effectively abolish their binding sites (Fig. 2B and C). To further verify whether both the 3′-UTR of PTEN and PRAP1 could be specifically targeted by miR-125a-3p, the luciferase plasmids were constructed for luciferase assays. As shown in Fig. 2D, when the HEK-293T cells were co-transfected with wt PTEN-3′-UTR and miR-125a-3p mimics, the luciferase activity had no difference with NC and mut PTEN-3′-UTR+mimics groups, indicating miR-125a-3p could not directly block 3′-UTR of PTEN, but regulate its level through other pathways. Meanwhile, the luciferase activity of wt PRAP1-3′-UTR+miR-125a-3p mimics group was obviously decreased in comparison to the NC and mut PRAP1-3′-UTR+mimics groups (Fig. 2E). Our findings suggested that miR-125a-3p directly suppressed the PRAP1 expression via specifically target its 3′-UTR.
 These target genes were initially verified by qPCR. (B and C) Both 3′-UTR of wild-type PTEN and PRAP1 contained a seven-nucleotide binding site (5′-UCACCUG-3′) of miR-125a-3p. (D and E) Dual-luciferase reporter assays performed to further verify the most probable targets of miR-125a-3p. Each value represents mean±SEM (n=3). β-Actin was considered as an internal control. *p<0.05 vs. Control group.")
MiR-125a-3p could specifically target the 3′-UTR of PRAP1. To investigate the molecular mechanism of the functional effects of miR-125a-3p on the hepatocyte proliferation, CKAP4, PTEN, PRAP1, FBXO31, P53, and SENP2 were predicted as the potential targets of miR-125a-3p by Targetscan and OUGene. (A) These target genes were initially verified by qPCR. (B and C) Both 3′-UTR of wild-type PTEN and PRAP1 contained a seven-nucleotide binding site (5′-UCACCUG-3′) of miR-125a-3p. (D and E) Dual-luciferase reporter assays performed to further verify the most probable targets of miR-125a-3p. Each value represents mean±SEM (n=3). β-Actin was considered as an internal control. *p<0.05 vs. Control group.
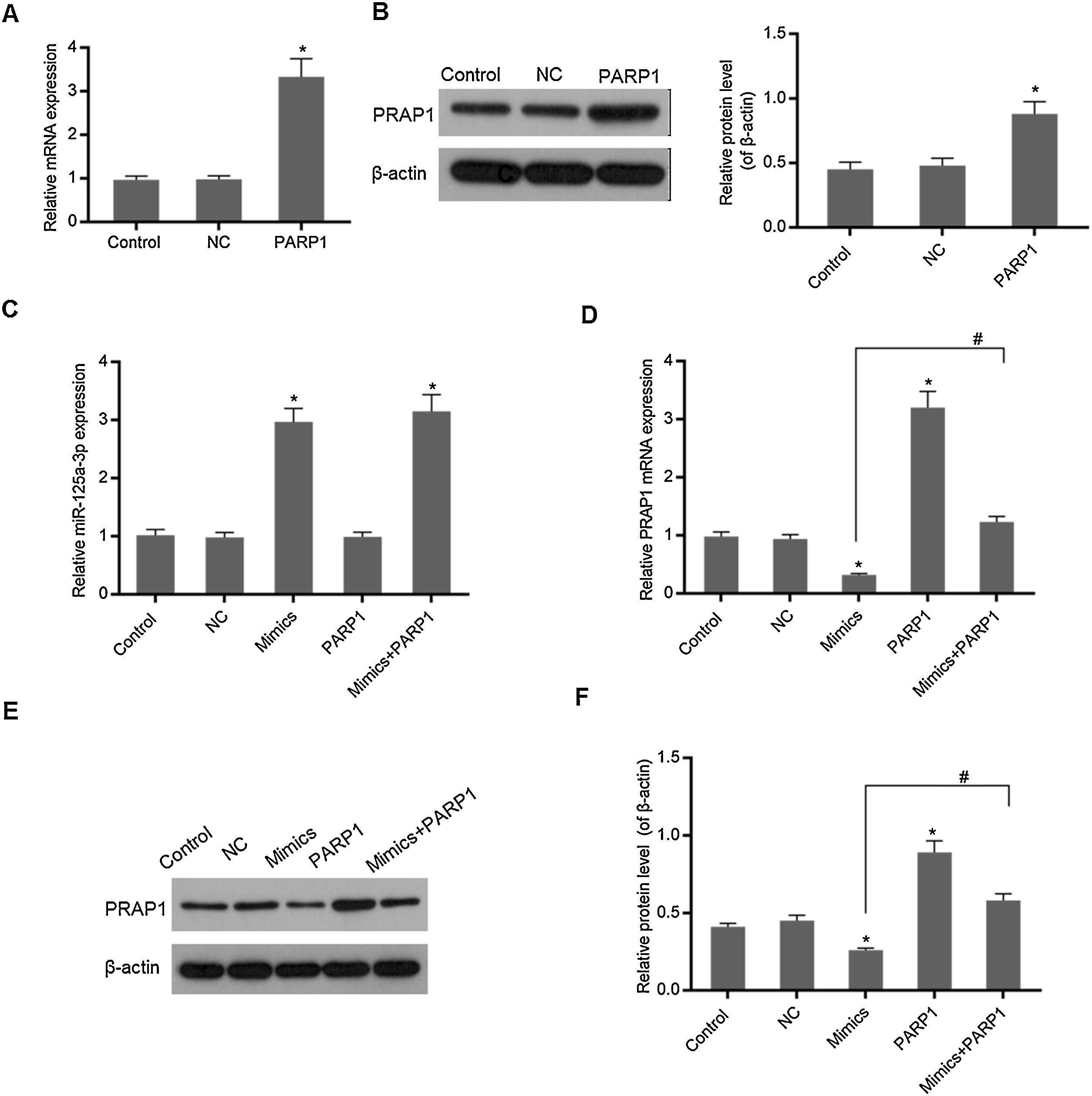
In Fig. 3A and B, overexpression PRAP1 vector has stable and efficiently expressed in the transfected cells. When the PRAP1 overexpression vector was co-transfected with miR-125a-3p mimics, the expression level of miR-125a-3p was almost unchanged (Fig. 3C). However, the mRNA and protein levels of PRAP1 were significantly upregulated, even under the control of miR-125a-3p (p<0.05, Fig. 3D–F). Therefore, the co-transfection of PRAP1 overexpression vector could contribute greatly to reversing the negative effects of miR-125a-3p.
 The transfection efficiency was measured by qPCR and Western blot. (C) The miR-125a-3p level was detected under the combined effects of miR-125a-3p mimics and PRAP1 overexpression vectors. (D–F) The mRNA and protein levels of PRAP1 were measured by qPCR and Western blot. Each value represents mean±SEM (n=3). U6 was set as the internal control for miR-125a-3p levels and β-actin was considered as an internal control for PRAP1. *p<0.05 vs. Control group; #p<0.05 vs. Mimics groups.")
PRAP1 overexpression could reverse the control of miR-125a-3p to its expression. PRAP1 was a considered target of miR-125a-3p. PRAP1 overexpression vector was constructed and co-transfected with miR-125a-3p mimics. (A and B) The transfection efficiency was measured by qPCR and Western blot. (C) The miR-125a-3p level was detected under the combined effects of miR-125a-3p mimics and PRAP1 overexpression vectors. (D–F) The mRNA and protein levels of PRAP1 were measured by qPCR and Western blot. Each value represents mean±SEM (n=3). U6 was set as the internal control for miR-125a-3p levels and β-actin was considered as an internal control for PRAP1. *p<0.05 vs. Control group; #p<0.05 vs. Mimics groups.
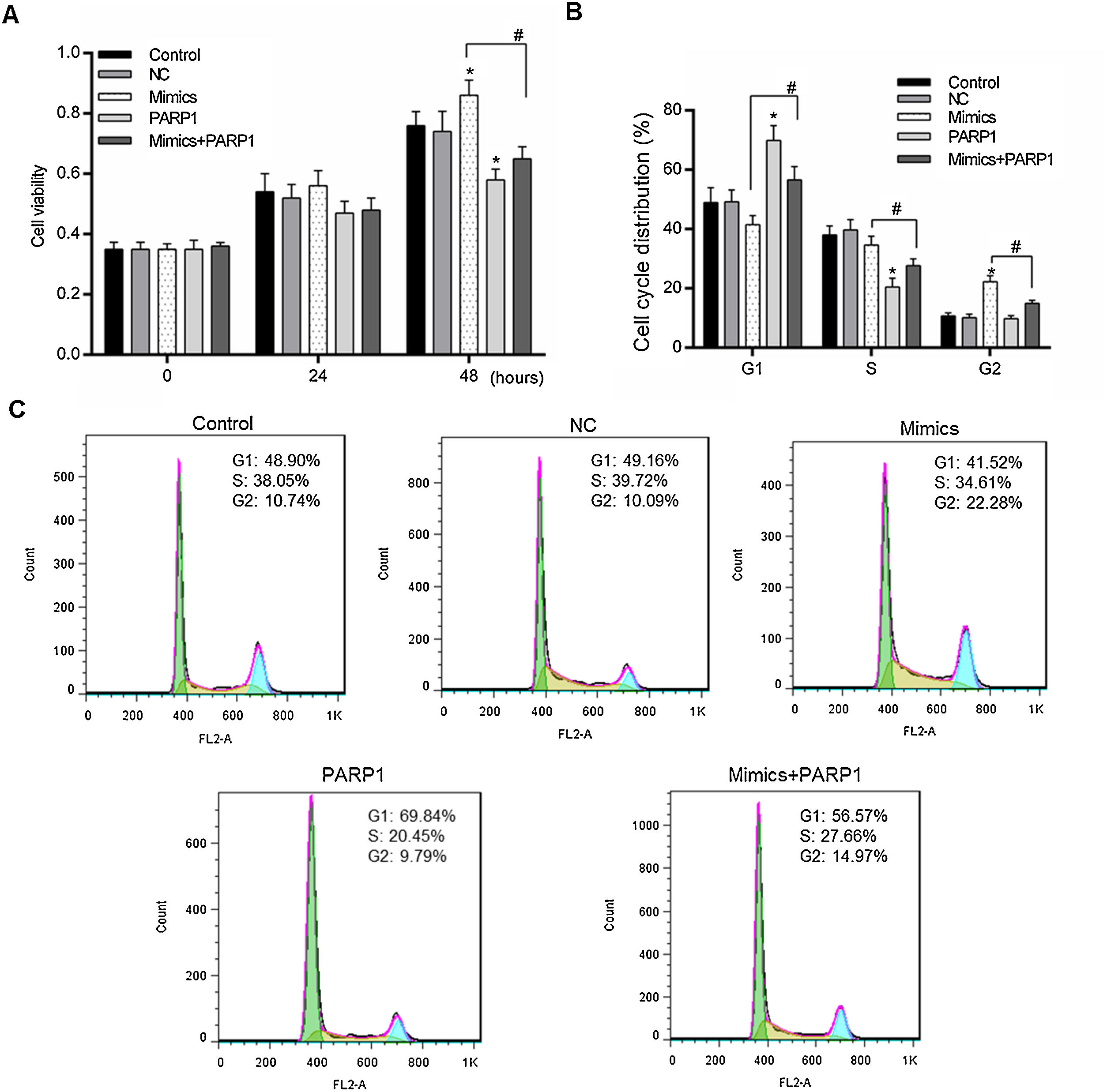
We further detected the HL-7702 cell viability and cell cycle distribution when PRAP1 was co-transfected with miR-125a-3p mimics. As previously described, miR-125a-3p positively associated with the HL-7702 cell viability after incubation for 48h, however, the cell viability in mimics+PRAP1 group was markedly suppressed in comparison to the Mimics group (p<0.05), even slightly lower than the control and NC group (Fig. 4A). In the meantime, the cell cycle assay showed that, after co-transfection with PRAP1 overexpression vector, the number of G1 phase HL-7702 cells was notably increased and the cell number at S and G2 phase was significantly decreased (p<0.05), indicating PRAP1 overexpression, similar with functional effect of miR-125a-3p inhibition, inhibited cell cycle progression through inducing cell cycle G1 phase arrest (Fig. 4B and C). Taken together, PRAP1 overexpression could effectively abolish positive effects of miR-125a-3p on HL-7702 cell proliferation and cell cycle progression.
 The cell viabilities of each experiment group cells were detected by CCK-8 kit each 24h. (B and C) The transfected cells were collected for the analysis of cell cycle distribution by flow cytometry. Each value represents mean±SEM (n=3). *p<0.05 vs. Control group; #p<0.05 vs. Mimics groups.")
PRAP1 overexpression could reduce the proliferation and cell cycle progression effects by miR-125a-3p on HL-7702 cells. (A) The cell viabilities of each experiment group cells were detected by CCK-8 kit each 24h. (B and C) The transfected cells were collected for the analysis of cell cycle distribution by flow cytometry. Each value represents mean±SEM (n=3). *p<0.05 vs. Control group; #p<0.05 vs. Mimics groups.
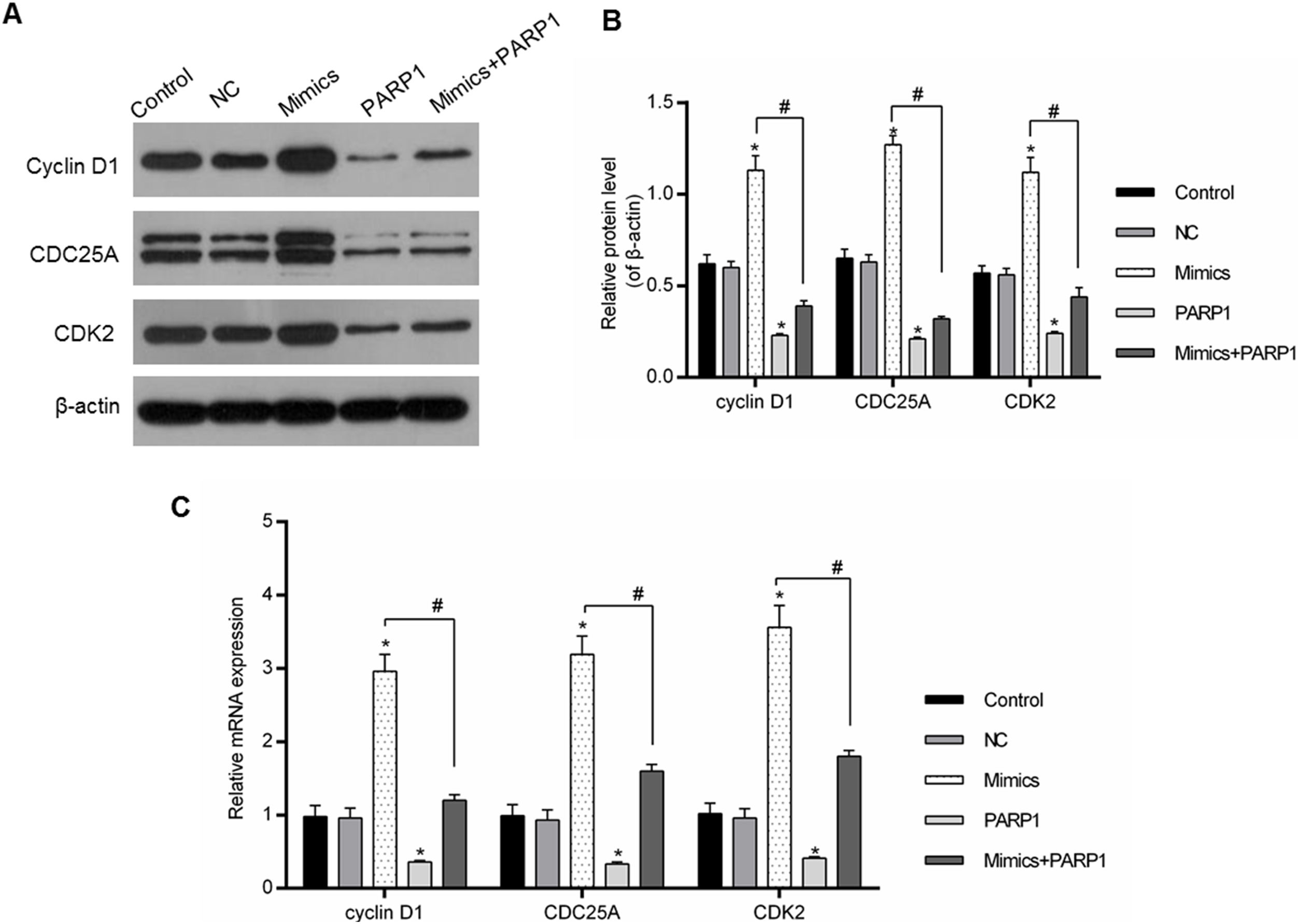
The cell cycle progression was driven by a variety of genes, therefore the regulation of cell-cycle-associated genes could directly affect the cell cycle progression. In our study, the expression levels of cyclin D1, CDC25A and CDK2 were chosen for the assessment of the PRAP1 influences and miR-125a-3p on the cell cycle progression. In Fig. 5A–C, miR-125a-3p mimics could markedly upregulate their mRNA and protein levels in comparison to the control and NC groups (p<0.05), while their expressions in PRAP1 overexpression group was obviously decreased (p<0.05). More importantly, when co-transfection with PRAP1 and miR-125a-3p, the expressions of cyclin D1, CDC25A, and CDK2 upregulated by miR-125a-5p was abolished by overexpressed PRAP1 (p<0.05). Therefore, PRAP1 overexpression inhibited the HL-7702 cell proliferation and cell cycle progression through reversing the positive effects of miR-125a-3p.
 expressions were examined by qPCR and Western blot. (A and B) MiR-125a-3p overexpression could obviously increase their protein levels, but PRAP1 co-transfection partially reversed the upregulated protein levels of cyclin D1, CDC25A, and CDK2 by miR-125a-3p mimics. (C) The changes in mRNA levels of these three genes were consistent with their protein levels. Each value represents mean±SEM (n=3). β-Actin was considered as an internal control. *p<0.05 vs. Control group; #p<0.05 vs. Mimics groups.")
PRAP1 overexpression reversed the auxo-action of miR-125a-3p in the expressions of cell-cycle-associated genes. Several cell cycle-associated gene (cyclin D1, CDC25A, and CDK2) expressions were examined by qPCR and Western blot. (A and B) MiR-125a-3p overexpression could obviously increase their protein levels, but PRAP1 co-transfection partially reversed the upregulated protein levels of cyclin D1, CDC25A, and CDK2 by miR-125a-3p mimics. (C) The changes in mRNA levels of these three genes were consistent with their protein levels. Each value represents mean±SEM (n=3). β-Actin was considered as an internal control. *p<0.05 vs. Control group; #p<0.05 vs. Mimics groups.
In this paper, our results revealed the increasing miRNA-125a-3p level could enhance the cell viability of hepatocytes, and promote the cell cycle progression through upregulating the expression of cyclin D1, CDC25A, and CDK2. More importantly, miR-125a-3p could specifically target PRAP1, while the co-transfection with PRAP1 overexpression could effectively reverse the auxo-action of miR-125a-3p in hepatocyte proliferation and cell cycle progression. Collectively, it is rational to think that miR-125a-3p overexpression positively affected cell cycle progression and promoted hepatocyte proliferation through targeting PRAP1 expression, and the elevated proliferative capacity of hepatocyte contributed to the liver regeneration process.
PRAP1 is initially described as a differential expression gene in the pregnant uterus of rodents [16], and it has been detected in several human tissues, including the gastrointestinal tract, the epithelium of the liver and kidneys [17]. It has been demonstrated that, once DNA damage appeared, for repairing the damage, cells in mammalian would induce cell-cycle arrest through promoting the activation of p53 pathway [18,19]. Recent studies showed that the mechanism of cell-cycle arrest mediated by p53 pathway in DNA damage cells mainly relied on the downstream gene activation, subsequently, these downstream genes, like p21, inhibited the expressions of CDK2 and CDK1, which were essential for the progression of G1/S and G2/M transition [20,21]. Currently, the positive effects of PRAP1 on DNA damage have been greatly proved [22]. In 2012, Huang et al. proved the functional effects of p53 on cell repair depended on cell cycle arrest mediated by p53 pathway, and PRAP1 expression played a vital role the induction of cell cycle arrest [23]. In our study, we observed that the number of G1 phase cells was much higher with the expression of PRAP1 increase, indicating that the elevated PRAP1 level may suppress the proliferation of hepatocytes through effectively inhibiting the cell cycle G1/S transition. In addition, previous research also demonstrated that PRAP1 was a novel mitotic arrest deficient 1 (MAD1) interacting partner, which was an essential component in mitotic checkpoint signaling [24,25], and the overexpression of PRAP1 could significantly decrease the protein level of MAD1, and ultimately result in an obvious downregulation of mitotic indices and severe chromosomal instability [26]. Based on these researches, our data demonstrate that PRAP1 overexpression had a negative effect on the liver regeneration through inhibiting the progression of cell cycle G1/S transition and hepatocyte proliferation. Therefore, PRAP1 might be a promising target for the improvement of liver regeneration.
The anti-cancer effect of miR-125a-3p has been widely reported, such as miR-125a-3p could effectively attenuate liver cancer cell migration and invasion [27], and miR-125a-3p also limited the development of glioma via directly modulating the expression of neuregulin 1 (Nrg1) [28], but limited data was about the functional effects of miR-125a-3p on liver regeneration process. A recent study indicated a high association between miR-125a-3p and liver regeneration, therefore the effects of miR-125a-3p overexpression and inhibition on the hepatocyte proliferation were assessed by cell viability and cell cycle assays, and we observed that increasing miR-125a-3p promoted cell cycle progression of hepatocytes and enhanced the proliferative capacity. Interestingly, our study revealed that PRAP1 was a predicted target of miR-125a-3p, this may explain why miR-125a-3p could promote cell proliferation and cell cycle progression of hepatocytes. In addition, we also found that overexpressed PRAP1 could inhibit the expressions of cyclin D1, CDC25A, and CDK2. As we know that the cyclin-dependent kinases (CDKs) family occupied an important place in the regulation of cell cycle progression, among this family, CDK2 positively correlated with the G1/S transition, which contributed to DNA replication [29]. In the meantime, the expression of cyclin D1 and CDC25A could induce the activation of CDK2, otherwise, cyclin D1 also had the ability to phosphorylate the retinoblastoma tumor suppressor protein family (Rb) and activate E2F transcription factors, and ultimately promote G1/S transition [30,31]. In our study, miR-125a-3p overexpression could enhance the expressions of cyclin D1, CDC25A and CDK2 through blocking PRAP1 expression, subsequently, promoting hepatocyte cell cycle progression.
In summary, our data firstly revealed the molecular mechanism of the functional effects of miR-125a-3p on the hepatocyte proliferation and liver regeneration. Firstly, PRAP1 expression could induce the cell cycle arrest at the G1/S transition, and inhibit hepatocyte proliferation and prevent the process of liver regeneration. Overexpressed miR-125a-3p could effectively improve the hepatocyte growth, proliferation, and cell-cycle progression through directly silencing the negative effects of PRAP1. Our study provides a novel insight into the positive effects of miR-125a-3p on liver regeneration.AbbreviationsCCK-8 cell counting kit-8 real-time quantitative PCR MicroRNAs proline-rich acidic protein 1 cyclin-dependent kinase 2 cell division cycle 25A interleukin-6 tumor necrosis factor alpha untranslated regions tyrosine-protein kinase Fyn non-small-cell lung cancer Dulbecco's modified eagle medium fetal bovine serum optical density mitotic arrest deficient 1 neuregulin 1 retinoblastoma tumor suppressor protein family cyclin-dependent kinases
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestThe authors declare no conflicts of interest.



