





The obesity pandemic that affects the global population generates one of the most unfavorable microenvironmental conditions in the hepatocyte, which triggers the metabolic hepatopathy known as non-alcoholic fatty liver; its annual rates increase in its prevalence and does not seem to improve in the future. The international consortia, LITMUS by the European Union and NIMBLE by the United States of America, have started a race for the development of hepatic steatosis and steatohepatitis reliable biomarkers to have an adequate diagnosis. MicroRNAs have been proposed as diagnostic and prognostic biomarkers involved in adaptation to changes in the liver microenvironment, which could improve clinical intervention strategies in patients with hepatic steatosis.
non-alcoholic fatty liver disease
non-alcoholic steatohepatitis
sterol regulatory element-binding protein 1
genome-wide association studies
single nucleotide polymorphisms
patatin like phospholipase domain containing protein 3
microRNAs
primary miRNA
double-stranded RNA binding protein DiGeorge critical region 8
transactivation response element RNA-binding protein
argonaute protein2
RNA-induced silencing complex
alanine aminotransferase
aspartate aminotransferase
cytokeratin-18
ATP binding cassette transporter member 1 of human transporter sub-family
3-hydroxy-3-methylglutaryl-coenzyme A reductase
fatty acid-binding protein 7
phosphatase and tensin homolog
peroxisome proliferator-activated receptor alpha
phosphoenolpyruvate carboxykinase
glucose-6-phosphatase
hepatocyte nuclear factor 4 alpha
nuclear factor kappa-beta
interleukin 6
tumor necrosis alpha factor
chemokine (C-C motif) ligand 2
hypoxia-inducible factor 1-α
mitogen-activated kinase 1
hepatitis B virus
hepatitis C virus
Non-alcoholic fatty liver disease (NAFLD) is defined as the presence of steatosis in at least 5% of hepatocytes [1] in the absence of other liver diseases such as viral hepatitis, autoimmune hepatitis, hemochromatosis, Wilson disease, significant alcohol consumption or for medication use. It is a complex clinical-pathological entity that arises from numerous genetic, environmental, behavioral, as well as social origin factors [2].
NAFLD is one of the most common liver diseases in the world, both in the adult and child population and number one in Western countries. Although more epidemiological studies are needed, compared to past decades, NAFLD prevalence increased from 2.8% to 46%, which has been associated with the global obesity and diabetes mellitus epidemic [3].
Ethnicity is a preponderant risk factor for NAFLD progression being the Latinos, one of the populations with the highest prevalence and risk, followed by Caucasians and then African Americans. Sex, in the same way, seems to be a risk determinant for NAFLD, being more frequent in men. The importance of knowing the risk factors associated with the disease lies in the fact that it follows an asymptomatic progression, and with few associated clinical complications. Approximately 5% of patients who showed NAFLD evidence was diagnosed with advanced non-alcoholic steatohepatitis (NASH), which is associated with cirrhosis mortality [4–6].
Several studies have shown that NAFLD prevalence and severity are proportionally related to the degree of obesity, with a strong association of 85% in obese subjects, 35% of patients with diabetes and up to 90% of patients with hyperlipidemia; in fact, it is reported that about 20–30% of obese patients progress to liver damage. On the other hand, Shida et al. demonstrated that a progressive reduction in muscle mass accompanied by an increase in visceral fat is associated with liver complications and fibrosis progression [7].
NAFLD is considered a hepatic manifestation of the metabolic syndrome and is closely related to central obesity, insulin resistance, glucose intolerance, and type 2 diabetes mellitus. About 20–70% of adult patients with NAFLD have type 2 diabetes mellitus, a decrease in insulin sensitivity, and an increase in muscle insulin resistance, white adipose tissue, which leads to liver fat accumulation. Dyslipidemia is another frequent finding in individuals with NAFLD, hypertriglyceridemia, hypercholesterolemia, or both are observed in up to 20–81% of patients. On the other hand, the complex interaction between circulating inflammatory mediators with organs and tissues, genetic background and some conditioning factors such as lifestyle (diet and physical activity), increase the risk and severity of NAFLD [8–10] (Fig. 1).
2Physiopathology NAFLD pathogenesis are the risk factors for the development of
NAFLD pathogenesis are the risk factors for the development of Under normal conditions, the liver is responsible for maintaining the balance between lipogenesis and β-oxidation of fatty acids; however, lipid homeostasis in NAFLD is altered. Patients with hepatic steatosis have insulin resistance; therefore, adipose tissue releases free fatty acids, which is a determining factor for lipid accumulation in macro and microvesicles in more than 5% of hepatocytes, being distributed mainly in the perivenular region [9–11]. Another compensatory mechanism caused by insulin resistance is de novo lipogenesis in the liver, as a result of the sterol regulatory element-binding protein 1 (SRBEP-1) overexpression [12].
SRBEP-1 is the main isoform expressed in the liver and tissues involved in energy homeostasis and is activated by insulin, liver receptor X-α, endocannabinoid CB1 receptor, and suppressor of cytokine signaling 3, and inhibited by glucagon [13,14]. In murine models, SREBP-1 overexpression induces lipodystrophy development, insulin resistance, and hepatic steatosis, postulating that the intrahepatic lesion through positive regulation of the proapoptotic molecule Fas, which increases in non-alcoholic steatohepatitis (NASH), and it is suggested that it plays an important role in apoptosis [15].
3HistologyThe range of histological abnormalities found in the NAFLD varies from simple steatosis to cirrhosis, each with characteristics that involve different cellular elements, as well as alterations in them. Simple liver steatosis is characterized by the presence of macrovesicular lipid droplets, usually diffuse and is not accompanied by fibrosis, unlike non-alcoholic steatohepatitis that is characterized by macro and microvesicular steatosis, in addition to ballooning degeneration, portal inflammatory infiltrate and perisinusoidal or portal fibrosis [11]. Hepatic cirrhosis is the last stage of NAFLD progression, with a chronic and evolutionary behavior that presents multiple complications, reducing life expectancy. This disease is defined by hepatocellular death, parenchyma replacement with fibrotic tissue, regenerative nodules, as well as liver function loss [16,17].
4Genetic and epigenetic factorsDuring the last decade, considerable variability in the susceptibility of developing NASH among individuals with NAFLD has been seen. Technology progress has brought new ways of studying the genes behavior, epigenetic modifications, and mechanisms in NAFLD pathogenesis. As a complex and multifactorial disease, modification in genetics and epigenetics play an essential role in its natural history.
The candidate genes in families and twins association study, adopted by the Genome-Wide Association Study (GWAS), allows the identification of genes and single nucleotide polymorphisms (SNPs), which are potentially involved in the mechanisms associated with NAFLD development and progression; for example, the patatin like phospholipase domain containing 3 (PNPLA3), is significantly associated with intrahepatic fat content and with more progressive forms of NAFLD. The SNP rs738409 of the PNPLA3 gene (M148I) (also known as adiponutrin), is the best characterized and is associated with ethnicity (mainly Hispanic Americans), steatosis, portal, and lobular inflammation, the appearance of Mallory-Denk bodies, and fibrosis development [18]. The carriers of this SNP exhibit a more severe NAFLD, and higher fibrosis levels, which provides strong molecular evidence that liver steatosis is a progressive disease [18,19].
Whereas epigenetics explains the possible mechanisms through which the cell can express some genes and silence others, it also refers to inheritable changes in DNA and histones that do not involve SNPs but modify the structure and condensation of chromatin, by which affect gene expression and phenotype. The epigenetic modifications mainly include microRNAs (miRNAs), DNA methylation, and histone acetylation that shape their particular characteristics, its specific tissue-expression and the susceptibility to develop certain diseases.
This review collects miRNAs that correlate them with the severity of NAFLD, and allow exploring its potential as therapeutic targets and future challenges [20].
5miRNAs biogenesis and mechanism of actionmiRNAs are a class of short non-coding RNAs transcribed by RNA polymerase II that possess approximately 19 to 25 nucleotides and function as post-transcriptional regulators of gene expression [21]. The genes that give rise to miRNAs are evolutionarily conserved and can be located within introns or exons, encoding proteins or in intergenic areas. Most of them are oriented in parallel with their host gene, suggesting that it is transcribed simultaneously, while the remaining group is transcribed from intergenic regions or from genes that make up independent transcription units [22].
miRNAs come from a much larger transcript of about 70–90 nucleotides that suffer many maturation steps. A classic primary miRNA (pri-miRNA) consists of a hairpin structure with an imperfect complementary nucleotide sequence single-stranded RNA that forms a double helix if the system energy allows it to fold, and a 5’ free end protected by a 7-methylguanosine, and at the opposite end a poly adenine tail. The pri-miRNA is modified inside the nucleus by RNase III Drosha in conjunction with the double-stranded RNA binding protein DiGeorge critical region 8 (DGCR8), whose catalytic action is to give rise to a chain shorter than ∼70 nucleotides in length, called precursor miRNA (pre-miRNA) that is transported to the cytoplasm through the nuclear export complex exportin 5 with GTP-binding nuclear protein RAN. Already in the cytoplasm, the pre-miRNA is processed by Dicer and TAR RNA-binding protein (TRBP) near the termination loop, liberating a mature double-stranded miRNA of 17–25 base pairs. Afterward, the double-stranded miRNA is cleaved by an argonaute protein (AGO 2), leaving a single chain miRNA called the guide strand (the second strand degrades), to create the effector complex called RNA-induced silencing complex (RISC) [23].
miRNAs direct gene expression by binding to the target mRNA 3’-untranslated region (3’-UTR), which results in the mRNA degradation, in case the binding is less strong, it will act to inhibit its translation; as a consequence, some miRNAs are partially complementary to one or several mRNAs. Indeed, miRNA specificity is given by the complementarity sequence between the mRNA interaction sites and the nucleotide sequence at position 2–8 at the 5 ‘end, known as the “seed region” [23,24].
miRNAs regulate the gene expression of various cellular processes, such as differentiation, invasion, and cell death, by binding to a mRNA whose function is to inhibit the translation or induce the degradation of a protein [25]. In humans, miRNAs mainly inhibit protein translation from the target genes and only infrequently cause mRNA degradation [24].
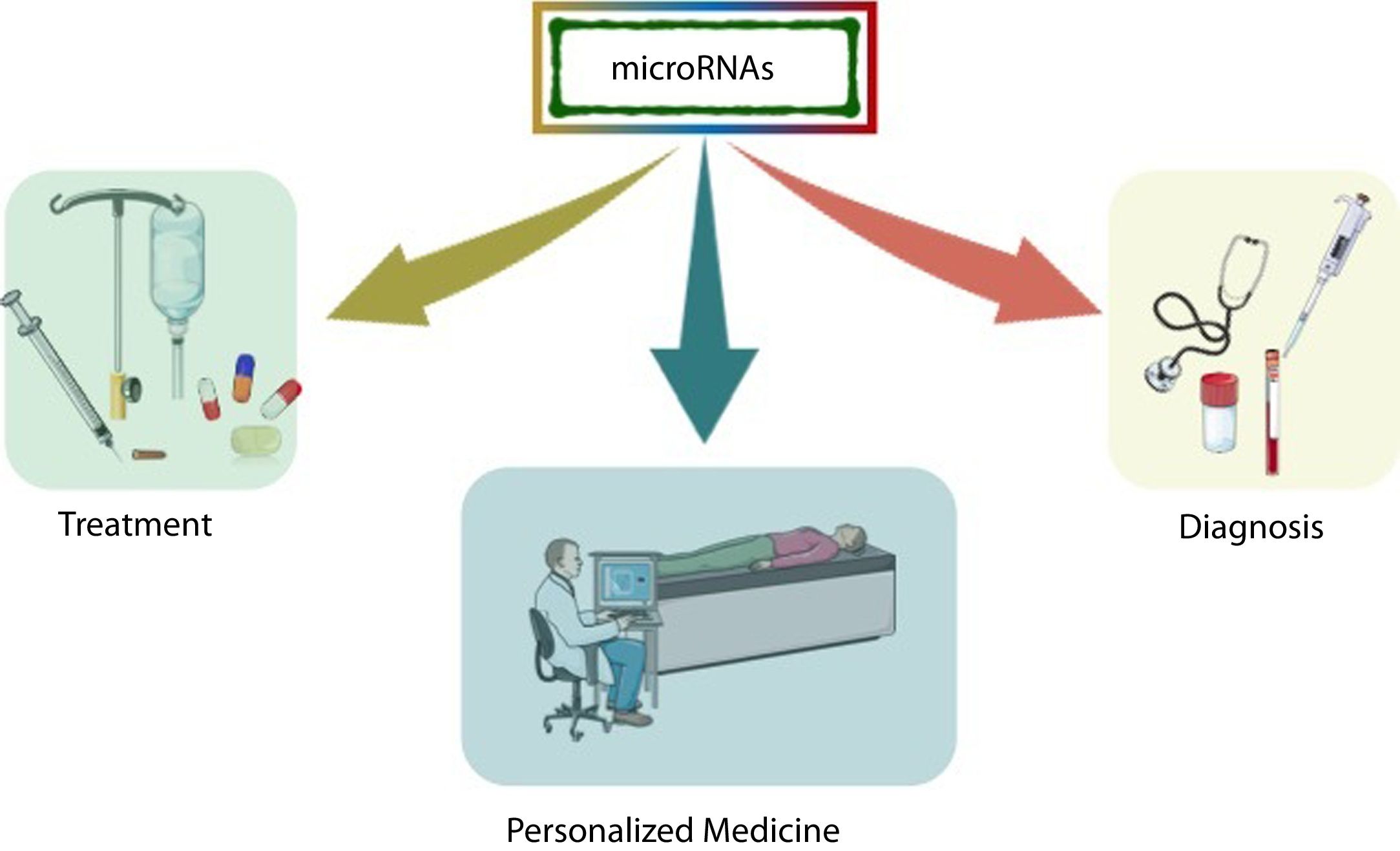
In the last decade, information regarding the number of human genes that are regulated by miRNAs has increased rapidly, and particular interest has been generated in determining the role of these molecules in various diseases, especially in the most complex ones that are subject to environmental influence. Because miRNAs play a crucial role in post-transcriptional control, they arise as critical therapeutic targets toward personalized medicine, where microarray research has allowed the identification of many miRNAs as biomarkers [26] (Fig. 2).
 miRNAs have been shown to play an important role in the development and progression of various pathologies, including
miRNAs have been shown to play an important role in the development and progression of various pathologies, including The targets that are recognized by miRNAs can be monogenic, polygenic (multifunctional), or even multiple miRNAs can target a single gene (redundancy), demonstrating that they have a sizeable regulatory capacity and a profound impact on health and disease. As well, miRNAs have been identified in serum, plasma, saliva, urine, and specific tissues, and are associated with proteins, lipids, and lipoproteins that make them more stable in the circulation [27].
Generally, miRNAs are bound to proteins, mainly AGO 2 or within exosomes, where they avoid degradation by the action of circulating RNAs [28,29]. The release to circulation can occur through a passive process during cell death or by cell microvesicles active release. For example, it has been shown that liver-specific miR-122 is found primarily in exosomes from alcoholic and non-alcoholic liver injury, while in toxic liver injury induced by paracetamol, most of the miR-122 is bound to proteins [30].
6Hepatic lipid metabolismSome essential miRNAs will be described as they address regulatory functions of hepatic lipid metabolism, in addition to their possible potential in NAFLD diagnosis and therapeutics. Fig. 3
 miRNAs have demonstrated a different expression pattern related to the different stages of hepatic steatosis.' title='Several
miRNAs have demonstrated a different expression pattern related to the different stages of hepatic steatosis.' title='Several The miR-122 is the most abundant and studied hepatic miRNA; it represents 70% of the total hepatic miRNAs [31] and is involved in hepatocyte proliferation and maturation by stimulating the expression of 24 specific genes, including the hepatocyte nuclear factor 6 (HNF6). It also interacts with numerous target genes involved in lipid and cholesterol metabolism [31–33]. Circulating levels of miR-122 correlate with alanine aminotransferase (ALT) levels in patients with NAFLD and be a better NAFLD severity indicator than classic liver function markers, including aspartate aminotransferase (AST) and cytokeratin-18 (CK18) [31,34].
Inhibition of miR-122 with antisense oligonucleotides in mice decreases hepatic fatty acids and cholesterol synthesis, as well as hepatic fatty acid oxidation, which diminishes plasma cholesterol levels [35,36]. Saturated fatty acids increase circulating miR-122 and reduce its levels in hepatocytes, which could be the result of increased miR-122 secretion [37,38].
On the other hand, miR-33a and miR33b are co-transcribed with the SREBP1 and SREBP2, which are regulators of de novo lipogenesis and cholesterol biosynthesis. They are also related to repression of the ATP binding cassette transporter member 1 of human transporter sub-family (ABCA1), which is essential for HDL synthesis through the regulation of ApoA1 and cholesterol binding [39]. Therefore, they contribute to the modulation of fatty acid metabolism pathways, cholesterol, and insulin synthesis. The inhibition of these miRNAs increases insulin sensitivity, β-oxidation, and HDL circulating levels, as well as the lipid accumulation reduction in arterial plaques, which is why they are proposed as potential therapeutic targets not only for NAFLD but also for metabolic syndrome management [40–42].
miR-21 is one of the best-studied miRNAs in the serum and liver of patients with NASH-fibrosis and hepatic carcinoma [43,44]. However, it has been mostly studied in in vivo and in vitro models. The activity of miR-21 induced by unsaturated fatty acids was increased in mice fed with a high-fat diet and human liver cells (HepG2); nevertheless, miR-21 elimination induces p53 transcription, which reduces the expression of genes that regulate lipogenesis and cell cycle arrest [45], but also the expression of several metabolic regulators, preventing glucose intolerance and steatosis [46]. Rodrigues et al. reported that miR-21 elimination in animals fed a high-fat diet supplemented with obeticholic acid decreased cholesterol accumulation, oxidative stress, and inflammation, which led them to develop minimal steatosis; in addition to restoring lipoprotein metabolism and hepatic insulin sensitivity [47].
In NAFLD, miR-21 regulates triglycerides, free cholesterol, and total cholesterol levels, which is achieved by 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) inhibition [48]. Also, the elevation of miR-21 inhibits fatty acid-binding protein 7 (FABP7), which notably induces fatty acid uptake and lipid accumulation [49]. miR-21 also targets phosphatase and tensin homolog (PTEN), which alters focal adhesion kinase phosphorylation and the expression of matrix metalloproteases 2 and 9, both mediators of cell growth, migration and invasion, which are phenotypic characteristics of cancer cells [50]; but also decrease peroxisome proliferator-activated receptor alpha (PPARα) expression that induce inflammation and fibrosis progression [51].
It has been shown that the decrease in mir-192 levels is associated with hepatic lipid accumulation induced by bisphenol A as a result of the positive regulation of SREBP1 due to defective DROSHA processing [52]. Also, miR-23b is capable of inhibiting Sirtuin 1 expression, which is a NAD-dependent deacetylase, resulting in TG deposition in the cytoplasm [53]. Besides, miR-26a inhibition increases cellular apoptosis and TG and cholesterol overload due to an increase in its metabolism [54]; therefore, these miRNAs have a vital function in lipid metabolism and the progression of NAFLD in human HepG2 cells. Xu et al. demonstrated that miR-34a inhibits hepatic VLDL secretion by promoting steatosis through interaction with HNF4α in patients with NASH and mice fed with HFD [55]. It has also been demonstrated in a murine model that the extract of Gynostemma pentaphylla (Thunb.) Makino lowers liver triglycerides and gut microbiota composition, particularly the phylum Firmicutes by miR-34a downregulation, which control the mRNA and protein levels of the target genes HNF4α, SIRT1, and PPARα[56].
On the other hand, miR-451a is capable of regulating the thyroid hormone response protein 14 expression, which has an essential role as a negative regulator of de novo lipogenesis [57]; also in Hepa1-6 cells, the miR-378 exerts an adverse action on Nrf-1 expression, which regulates lipid metabolism [58] miR-375 inhibition increases adiponectin expression, prevents the intracellular lipid accumulation and decreases the levels of leptin and inflammatory cytokines [59]. miRNAs, miR-190b [60], miR-27ª [61], miR-194 [62], and miR-181b [63] have demonstrated their possible role as negative protein regulators involved in NAFLD development, either by insulin sensitivity decrease or through de novo fatty acids synthesis.
7Insulin resistanceCurrently, several miRNAs that modulate glucose metabolism, as well as insulin resistance, have been reported. miR-29 family and miR-122 are linked through metabolic pathways by regulating insulin resistance related to NAFLD [64]. Also, in patients with insulin resistance and NAFLD, miR-33 inhibits gluconeogenesis by the activation of phosphoenolpyruvate carboxykinase (PCK1) and glucose-6-phosphatase (G6PC), which are two essential enzymes for glucose biosynthesis and homeostasis [65].
On the other hand, the miR-375 regulates glucose homeostasis and is responsible for pancreatic beta cells responding to insulin resistance. However, some reports associate it with elevated levels in NASH patients when compared to patients with simple steatosis [59]. The miRNA-30b is elevated in rats with insulin resistance and NAFLD, and it was demonstrated that it is a sarco(endo)plasmic reticulum Ca2+-ATPase 2b regulator [66].
8Inflammatory pathwaysNAFLD entails significant cellular stress that leads to the activation of inflammatory pathways. The miR-34a is highly expressed in patients with type 2 diabetes mellitus, liver steatosis and NASH is the best-characterized Sirtuin 1 (SIRT1) regulator, and is highly sensitive to hepatic lipid overload that modulates oxidative stress and lipid metabolism. miR-34a silencing restores SIRT1 expression and the peroxisome proliferator-activated receptor alpha (PPARα), which results in steatosis improvement [67]. On the contrary, the induction of miR-34a is related to hepatocyte nuclear factor 4 alpha (HNF4α) inhibition in patients with NAFLD and as a pro-inflammatory molecule through the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [55].
Moreover, miR-155 is a pro-inflammatory miRNA that appears to control innate and adaptive immunity in NAFLD, since it is elevated in hepatocytes and Kuppfer cells of in vivo models fed a methionine deficient diet [68,69]. Also, a study in mice fed a high-fat diet with lean and obese phenotypes demonstrated that miR-140 loss results in increased TLR-4 expression, and consequently, a greater inflammatory activity through the TLR4/NFκB pathway, this signaling axis promotes NAFLD progression and fibrosis development [70].
On the other hand, miR-223 was first described as a modulator of hematopoietic differentiation and has been proposed as part of the master set that regulates the inflammasome function. Recent studies have shown that miR-223 expression is deregulated in several liver diseases, including viral hepatitis infections, alcohol or drug-induced liver damage, non-alcoholic fatty liver disease, cirrhosis, and hepatocellular carcinoma [70].
9miRNAs that regulate NAFLD progressionmiRNAs deregulation in NAFLD could prepare hepatocytes for malignancy, either directly or indirectly.
Pirola et al described that miR-122 levels are 7.2 times higher in patients with NASH compared to healthy subjects and 3.1 times higher compared to patients with simple steatosis, proposing it as an extrahepatic NASH marker [31]. However, there is contrary data to those described by Pirola, which report a negative correlation between hepatic and serum miR-122 levels. In NASH, miR-122 expression decreases ten times compared to the levels of patients with simple steatosis [71,38]. In fact, in animal models, miR-122 serum levels correlate positively with NAFLD severity, even in the absence of ALT elevation. Indeed, liver-specific miR-122 knockout mice rapidly develop NASH, due to increased lipogenesis, alterations in lipid secretion, interleukin 6 (IL-6), tumor necrosis alpha factor (TNF-α) and chemokine (C-C motif) ligand 2 (CCL2) elevation, but also macrophage recruitment. On the other hand, miR-122 reduction induces fibrogenic pathways expression by promoting the hypoxia-inducible factor 1-α (HIF1α) and mitogen-activated kinase 1 (MAPK1), which can also induce the development of hepatocellular carcinoma [72]. As well, mice with suppressed miR-122 also showed lower levels of serum triglycerides and cholesterol; however, increased circulating levels of miR-122 have also been detected in patients with diseases such as viral hepatitis B and C (HBV, HCV), as well as alcohol and drug-induced liver disease [73–75].
Another important point is that miR-33a is strongly expressed in activated star cells, and their concentrations correlate with TGF-β-induced expression of type I collagen, suggesting its participation in the development of fibrosis [42,76]. Indeed, miR-34a also has pleiotropic functions in the proliferation regulation, differentiation, and programmed cell death; it is one of the most augmented during hepatic fibrogenesis in both animal and human models [77,78].
The miR-21 can modulate fibrogenesis favoring the activation of Akt [47]. However, miR-21 has been proposed to play a role in steatosis progression to hepatocarcinoma by inhibiting the transcription factor HMG-Box 1 (HBP1) [79]. As well, miR-192 is expressed in inactive stellar cells, which favors inhibition, activation, proliferation, and migration; however, it plays an important role in fibrosis development induced by transforming growth factor beta-1 (TGFβ1), as in the SMAD signaling activation [80]. Like miR-122, the serum concentration of miR-192 is elevated, while in liver tissue, it is negatively regulated in patients with NASH compared to patients with simple steatosis; therefore, it could be proposed as a strong prognosis biomarker [80,34].
On the other hand, miR-16 is constituted by a miRNAs superfamily, which includes miR-16, miR-497, miR-195, miR-322 and miR-15, particularly the miR-15 and miR-16 members have been proposed as hepatic fibrosis and hepatocarcinogenesis regulatory molecules [81]. Higher circulating levels of miR-16 were reported in patients with NAFLD compared to healthy controls, and their levels correlated with the disease severity, supporting its use as NAFLD staging biomarkers. miR-16 deregulation can promote the stellar cells activation by regulation of the guanine nucleotide signaling that binds to subunit 12 (Gα12), a key transducer of G protein-coupled receptors [81].
Ogawa et al reported the first evidence that miR-221 and miR-222 are elevated in patients affected by NASH in a fibrosis-dependent manner; but also showed that mice with thioacetamide or a methionine-deficient diet, miR-221/222 increased levels activate stellate cells, as well as collagen type 1 alpha 1, and anti-alpha smooth muscle actin expression, inducing fibrosis compared to controls [82]. Currently, miR-221/222 are considered oncomiRs and are under study for their potential in the prediction and induction of tumorigenesis [83].
10ConclusionIn summary, microRNAs perform critical functions during development and cellular homeostasis by modulating the expression of various genes involved in critical biological processes, such as differentiation, proliferation, and cell death. The role of miRNAs as regulatory molecules of different critical mechanisms for the development of NAFLD, such as insulin sensitization and the post-receptor alterations that this entails, as well as the regulation of the expression of crucial proteins both in the de novo synthesis as an export of intrahepatic fatty acids has been demonstrated in different experimental models that future studies should look for ways to determine the genetic signatures of the different stages of NAFLD. MicroRNA expression signatures have consistent and reproducible levels and stability in human peripheral blood that make them quantifiable, thereby making them potential biomarkers for clinical diagnosis and potential therapeutic targets.
Conflict of interestGuillermo Nahúm López-Sánchez, Mayra Dominguez-Pérez, Norberto Carlos Chávez-Tapia, Misael Uribe and Natalia Nuño Lámbarri declare that they have no conflict of interest.
We appreciate the support of Medica Sur Clinic & Foundation.
We confirm that this work is original and has not been published nor is it currently under consideration for publication elsewhere, in whole or in part, and we have not had any competing financial interests or commercial relationships that might pose a conflict of interest.



