



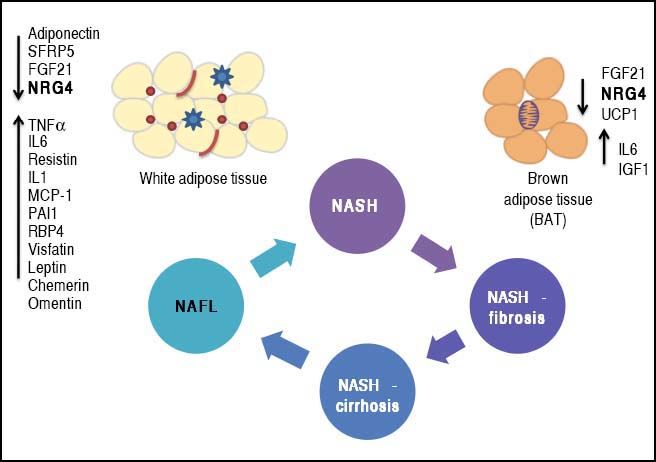

Phenotypic modulation of NAFLD-severity by molecules derived from white (adipokines) and brown (batokines) adipose tissue may be important in inducing or protecting against the progression of the disease. Adipose tissue-derived factors can promote the progression of NAFLD towards severe histological stages (NASH-fibrosis and NASHcirrhosis).
This effect can be modulated by the release of adipokines or batokines that directly trigger an inflammatory response in the liver tissue or indirectly modulate related phenotypes, such as insulin resistance. Metabolically dysfunctional adipose tissue, which is often infiltrated by macrophages and crown-like histological structures, may also show impaired production of anti-inflammatory cytokines, which may favor NAFLD progression into aggressive phenotypes by preventing its protective effects on the liver tissue.
Opinion Article on the MS published in the Journal of Clinical Investigation entitled: Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression.
Nonalcoholic fatty liver disease (NAFLD) is regarded as the most prevalent chronic liver disease worldwide.1 Although the disease is considered benign and having a relatively good prognosis when the diagnosis is made at earlier histological stages, the progression into severe clinical forms, including nonalcoholic steatohepatitis (NASH), NASH-fibrosis and NASH-cirrhosis, imposes tremendous medical challenges.1 For these reasons, extensive body of research aimed at the search for factors and or causes that modify the natural history of NAFLD is currently being conducted.
The complexity of the NAFLD-associated clinical picture, specifically the association with co-morbidities such as type 2 diabetes (T2D),1 obesity and cardiovascular disease,2–4 suggests the presence of an even more intricate network of pathogenic mechanisms that not only interact with each other but also increase the chances of having a more dramatic and severe phenotype. In this regard, the cross-talk between NAFLD and adipose tissue has gained particular attention of researchers and practitioners because cytokines derived from either white fat-WAT (adi-pokines) or brown fat-BAT (batokines) play a remarkable role in the development and maintenance of NAFLD disease severity. Figure 1 illustrates the concept of phenotypic modulation of NAFLD severity by molecules derived from adipose tissue that act as paracrine and endocrine hormones.5,6
 and brown (batokines) adipose tissue by inducing or protecting against the progression of the disease. Adipose tissue-derived factors can promote the progression of NAFLD towards severe histological stages (NASH-fibrosis and NASHcirrhosis). This effect can be modulated by the release of adipokines that directly trigger an inflammatory response in the liver tissue or indirectly modulate related phenotypes, such as insulin resistance (up-arrows). Metabolically dysfunctional adipose tissue, which is often infiltrated by macrophages and crown-like histological structures, may also show impaired production of anti-inflammatory adipokines, which may favor NAFLD progression into aggressive phenotypes by preventing its protective effects on the liver tissue (down-arrows). SFRP5: secreted frizzled-related protein 5. UCP1: uncoupling protein 1. FGF21: fibroblast growth factor 21. TNF a: tumor necrosis factor a. IL1 and IL6: interleukin 1 and 6. MCP-1: mono-cyte chemotactic protien-1.")
Phenotypic modulation of NAFLD-severity by molecules derived from white (adipokines) and brown (batokines) adipose tissue by inducing or protecting against the progression of the disease. Adipose tissue-derived factors can promote the progression of NAFLD towards severe histological stages (NASH-fibrosis and NASHcirrhosis). This effect can be modulated by the release of adipokines that directly trigger an inflammatory response in the liver tissue or indirectly modulate related phenotypes, such as insulin resistance (up-arrows). Metabolically dysfunctional adipose tissue, which is often infiltrated by macrophages and crown-like histological structures, may also show impaired production of anti-inflammatory adipokines, which may favor NAFLD progression into aggressive phenotypes by preventing its protective effects on the liver tissue (down-arrows). SFRP5: secreted frizzled-related protein 5. UCP1: uncoupling protein 1. FGF21: fibroblast growth factor 21. TNF a: tumor necrosis factor a. IL1 and IL6: interleukin 1 and 6. MCP-1: mono-cyte chemotactic protien-1.
Pathways involved in organ damage and injury, including cellular apoptosis or other process associated with cell death, as well as persistent tissue inflammation and the consequent fibrogenesis, are all triggered by multiple factors, including genetic predisposition and epigenetic modifiers.7–9 Nevertheless, the exact mechanism/s that promote the transition of NAFL (nonalcoholic fatty liver) to NASH remain(s) elusive.
Guo, et al. recently reported the role of Neuregulin 4 (Nrg4), an adipose tissue-enriched endocrine factor, in the development of NASH.10 Specifically, the authors used a mouse model of high-fat-fructose diet-induced NASH that recapitulates main histological features of human NASH.10 Key molecular findings associated with the presence of NASH in mice were:
- •
Cell death, which was associated with both increased phosphorylation of the member of the MAP-kinase family JNK1/2 and reduced protein levels of the inhibitor of apoptotic and necroptotic cell death c-FLIPL (Caspase 8 and FADD-Like Apoptosis Regulatory Protein, CFLAR), and
- •
Significantly decreased mRNA expression of Nrg4 in epididymal WAT and BAT.10
Subsequent Nrg4 gain- and loss-of-function studies in mouse models showed that restoration of Nrg4 signaling protects animals from NASH; specifically, Nrg4 signaling protects animals from stress-induced cell death through interaction with c-FLIP.10 Notably, genes associated with fibrogenesis (Col1a1, Acta2, Tgfb1, and Mmp13) and inflammation (Tnfa, Il1b, Il12b, Nos2, Ccl2, Ccl5, and Adgre1) were upregulated in mice lacking Nrg4, suggesting that Nrg4 deficiency exacerbated liver inflammation and fibrosis following NASH-diet feeding.10 It is worth noting that a similar profile in human NAFLD was described by our research group.2
Considering the aforementioned findings collectively, it is plausible to hypothesize that NAFLD progression is a process in which physiological mechanisms of tissue homeostasis collapse. There is compelling evidence from human studies supporting this hypothesis. For instance, ballooning degeneration —a hallmark histological feature of NAFLD severity and NASH progression— is associated with down-regulation of liver HSP27 gene and protein expression.11 HSP27 is a member of the heat shock family of proteins also known as stress-responsive protein 27. Furthermore, NASH development is associated with mitochondrial dysfunction, increased mitochondrial DNA (mtDNA) genetic diversity, down-regulation of liver expression of genes of the oxidative phosphorylation (OXPHOS) chain, and aberrant patterns of mtDNA methylation.7,8,12,13
The first question that immediately emerges is what factor/s trigger this complex picture?
Robust evidence supports the notion that NAFLD severity is associated with metabolic stress. In fact, it has been linked with increased transamination reactions that would promote coping with the liver metabolic derangement.14 This, in turn, leads to changes in the amounts of amino acids released into the circulation, particularly in pathways related to glutamic acid. 13,14 As reported recently, glutaminolysis controls accumulation of myofibroblast hepatic stellate cells.15 Deregulated liver metabolism also leads to aberrant patters of hedgehog signaling16,17 and insufficient ketogenesis, which result in extensive hepatocyte injury and inflammation, decreased glycemia, and deranged hepatic TCA (tricarboxylic acid) cycle intermediate concentrations.14,18
The second, and even more difficult to answer question is why a brown fat-derived secreted factor is important in maintaining liver tissue homeostasis?
It is well known that systemic metabolic homeostasis is regulated by the “in concert” action of a myriad of tissue-derived factors, which originate from different sources, including adipose tissue, gut, muscle, liver and bone. These hormones / growth factors / bioactive molecules basically sense global metabolism and energy homeostasis. These mechanisms may eventually fail to ensure cell / tissue physiological functions. Alternatively, they can simply malfunction because the affected cell / tissue has abnormally increased its metabolic demands, which might occur in common diseases like NAFLD and obesity, as well as in cancer. In the latter case, epidermal growth factors (EGFs) are critically implicated in cancer growth and survival by engaging a number of proteins via the ErbB (Tyrosine Kinase-Type Cell Surface Receptor HER2) family of proteins. In this regard, the neuregulins, including NRG4, activate type-1 growth factor receptors involved in initiating cell-to-cell signaling through tyrosine phosphorylation.19 Neuregulins transduce signals via activation of the ErbB receptors, specifically ErbB4.
Evidence supporting the role of Nrg4 in NAFLD and metabolic syndrome, while presently limited, is promising. For example, it has been shown that Nrg4 attenuates hepatic lipogenic signaling and preserves glucose and lipid homeostasis in experimental models of obesity.20 In this particular study, Wang, et al. demonstrated in mice that Nrg4 negatively regulates de novo lipogenesis mediated by Lxr (liver X receptor) and Srebplc (sterol regulatory element binding transcription factor 1) in a cell-autonomous manner.20
Data yielded by human studies is not only insufficient, but is also inconclusive. Kang, et al. found that the circulating NRG4 levels are increased in patients with type 2 diabetes and positively correlate with serum glucose level, HOMA-IR, and serum triglycerides;21 however, opposite results were reported by Yan, et al.22 Controversial evidence has also been derived from studies of gestational diabetes.23,24 For example, Cai, et al. reported that NRG4 might protect against metabolic syndrome development.25 Similarly, Dai, et al. found that patients with NAFLD also show reduced levels of serum NRG4.26
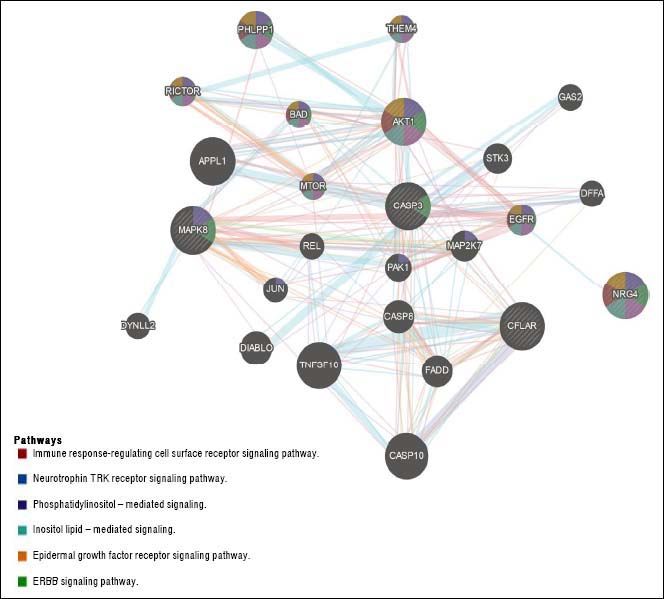
The question that still remains to be answered is whether the role of Nrg4 in protecting hepatocytes from apoptosis and necroptosis triggered by metabolic stress is mediated by a circulating endocrine signaling released from WAT or BAT, or if it is a truly liver-specific derived Nrg4. NRG4 human liver gene and protein expression levels are considerably low (http://www.proteinatlas.org/ ENSG00000169752-NRG4/tissue). Moreover, it is presently unclear whether Nrg4 signaling is mediated by interaction with hepatocytes or whether the actual effector/s of Nrg4 are non-liver related resident cells found in NAFLD and NASH. Hence, what exactly determines that Nrg4 is a key molecule in protecting the liver from severe NAFLD is uncertain. There are many possibilities, including the fact that Nrg4 indeed controls insulin sensitivity and secretion, as Nrg4 seems to be a potent insulin releaser.27 or macrophages migration into the liver. Still, Guo, et al. took the first steps toward unraveling the mechanisms involving the role of Nrg4 in protecting fatty liver from severe NASH. A list of Gene Ontology (GO) terms and biological process for NRG4 gene and its interaction network are shown in table 1 and figure 2, respectively. It is clear that this molecule seems to play a key role in ensuring effective protection against the progression of NAFLD by opposing caspase-mediated cell death. Interestingly, human “knock-out” for NRG4 has been identified; however, its phenotypic manifestation in the liver remains to be characterized.28
Gene Ontology (GO) - Biological Process for NRG4 Gene.
| GO ID | GO term. |
|---|---|
| GO:0000165 | MAPK cascade. |
| GO:0014066 | Regulation of phosphatidylinositol 3-kinase signaling. |
| GO:0018108 | Peptidyl-tyrosine phosphorylation. |
| GO:0035556 | Intracellular signal transduction. |
| GO:0038111 | Interleukin-7-mediated signaling pathway. |
| GO:0038128 | ERBB2 signaling pathway. |
| GO:0043547 | Positive regulation of GTPase activity. |
| GO:0046854 | Phosphatidylinositol phosphorylation. |
| GO:0048015 | Phosphatidylinositol-mediated signaling. |
| GO:0048513 | Animal organ development. |
| GO:1901185 | Negative regulation of ERBB signaling pathway. |
| GO:2000145 | Regulation of cell motility. |
NRG4 (Neuregulin 4) is a protein coding gene. Among its related pathways are RET signaling (REarranged during Transfection) and signaling by ERBB2. RET (Ret Proto-Oncogene) encodes one of the receptor tyrosine kinases, which are cell-surface molecules that transduce signals for cell growth and differentiation. ERBB2 (Erb-B2 Receptor Tyrosine Kinase 2) encodes a member of the epidermal growth factor (EGF) receptor family of receptor tyrosine kinases; this protein has no ligand binding domain of its own and therefore cannot bind growth factors. GO annotations related to NRG4 include receptor binding and growth factor activity.
. NRG4: neuregulin 4; CFLAR CASP8 and FADD like apoptosis regulator; MAPK8: mitogen-activated protein kinase 8. CASP3: caspase 3. AKT1: AKT serine/threonine kinase 1. APPL1: adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1. TNFSF10: tumor necrosis factor superfamily member 10. CASP10: caspase 10. PHLPP1: PH domain and leucine rich repeat protein phosphatase 1. DIABLO: diablo IAP-binding mitochondrial protein. CASP8: caspase 8. FADD: Fas associated via death domain. EGFR: epidermal growth factor receptor. STK3: serine/threonine kinase 3. MAP2K7: mitogen-activated protein kinase kinase 7. RICTOR: RPTOR independent companion of MTOR complex 2. PAK1: p21 (RAC1) activated kinase 1. BAD: BCL2 associated agonist of cell death. THEM4: thioesterase superfamily member 4. MTOR: mechanistic target of rapamycin. DFFA: DNA fragmentation factor subunit alpha. DYNLL2: dynein light chain LC8-type 2. GAS2: growth arrest specific 2. JUN: Jun proto-oncogene. AP-1 transcription factor subunit. REL REL proto-oncogene. NF- κBsubunit.")
NRG4 interaction network. Gene-gene functional network construction was done using GeneMANIA, available at:https://github.com/GeneMANIA/ genemania; visualization was performed by Cytoscape (http://apps.cytoscape.org/apps/GeneMania). NRG4: neuregulin 4; CFLAR CASP8 and FADD like apoptosis regulator; MAPK8: mitogen-activated protein kinase 8. CASP3: caspase 3. AKT1: AKT serine/threonine kinase 1. APPL1: adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1. TNFSF10: tumor necrosis factor superfamily member 10. CASP10: caspase 10. PHLPP1: PH domain and leucine rich repeat protein phosphatase 1. DIABLO: diablo IAP-binding mitochondrial protein. CASP8: caspase 8. FADD: Fas associated via death domain. EGFR: epidermal growth factor receptor. STK3: serine/threonine kinase 3. MAP2K7: mitogen-activated protein kinase kinase 7. RICTOR: RPTOR independent companion of MTOR complex 2. PAK1: p21 (RAC1) activated kinase 1. BAD: BCL2 associated agonist of cell death. THEM4: thioesterase superfamily member 4. MTOR: mechanistic target of rapamycin. DFFA: DNA fragmentation factor subunit alpha. DYNLL2: dynein light chain LC8-type 2. GAS2: growth arrest specific 2. JUN: Jun proto-oncogene. AP-1 transcription factor subunit. REL REL proto-oncogene. NF- κBsubunit.
In conclusion, there is substantial evidence suggesting that dysregulation of molecular mediators of metabolic homeostasis modulates the natural history of NAFLD. Both their upregulation and deficiency can either cause or exacerbate an adverse phenotype by evading from protective mechanisms against liver damage (Figure 1). Hence, the evolution of NAFLD into severe NASH is mediated by loss of systemic and/ or local protective mechanisms of organ damage.



