




Nonalcoholic steatohepatitis (NASH) is a form of fatty liver disease where benign hepatic steatosis leads to chronic inflammation in the steatotic liver of a patient without any history of alcohol abuse. Mechanisms underlying the progression of hepatic steatosis to NASH have long been investigated. This review outlines the potential role of peroxisomal dysfunctions in exacerbating the disease in NASH. Loss of peroxisomes as well as impaired peroxisomal functions have been demonstrated to occur in inflammatory conditions including NASH. Because peroxisomes and mitochondria co-operatively perform many metabolic functions including O2 and lipid metabolisms, a compromised peroxisomal biogenesis and function can potentially contribute to defective lipid and reactive oxygen species metabolism which in turn can lead the progression of disease in NASH. Impaired peroxisomal biogenesis and function may be due to the decreased expression of peroxisomal proliferator-activated receptor-α (PPAR-α), the major transcription factor of peroxisomal biogenesis. Recent studies indicate that the reduced expression of PPAR-α in NASH is correlated with the activation of the toll-like receptor-4 pathway (TLR-4). Further investigations are required to establish the mechanistic connection between the TLR-4 pathway and PPAR-α-dependent impaired biogenesis/function of peroxisomes in NASH.
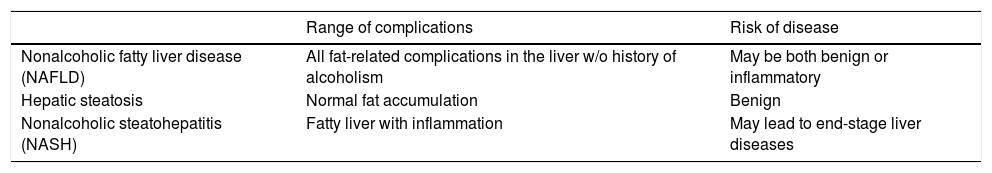
Nonalcoholic fatty liver disease (NAFLD) is defined as a condition with excessive fat accumulation and related complications in the liver of patients without any record of alcoholism [1]. NAFLD is an umbrella term which encompasses simple steatosis to nonalcoholic steatohepatitis (NASH) [2]. Hepatic steatosis refers to the normal fat accumulation in the liver without or with minor inflammation. Hepatic steatosis is benign, but longstanding steatosis can lead to the development of NASH which is the advanced stage of NAFLD showing a clear indication of chronic inflammation in the fatty liver [3,4] (Table 1). NAFLD is a rapidly growing health problem throughout the world. Currently, its global incidence is approximately 25% [5]. Of the individuals with NAFLD, approximately 7–30% meet the clinical criteria for NASH [6]. A major concern for individuals with NASH is the likelihood of their progression to end-stage liver diseases, that may eventually require transplantation [7]. NASH is projected to become the leading cause of liver-related morbidity and mortality superseding hepatitis C virus [8–11].
Nonalcoholic fatty liver disease, hepatic steatosis and nonalcoholic steatohepatitis.
| Range of complications | Risk of disease | |
|---|---|---|
| Nonalcoholic fatty liver disease (NAFLD) | All fat-related complications in the liver w/o history of alcoholism | May be both benign or inflammatory |
| Hepatic steatosis | Normal fat accumulation | Benign |
| Nonalcoholic steatohepatitis (NASH) | Fatty liver with inflammation | May lead to end-stage liver diseases |
Oxidative stress is considered as a key pro-inflammatory mediator of NASH [12]. Oxidative stress in hepatocytes can result from fat overload which is directly associated with an elevated level of circulating free fatty acids [13,14]. The concentration of circulating free fatty acids is low in a normal condition which is increased in metabolic syndrome [15]. Influx of elevated level of circulating free fatty acids into hepatocytes results in an increased fatty acid oxidation in mitochondrial and extra-mitochondrial sites leading to over-production of reactive oxygen species (ROS) [16]. Excess generation of ROS creates an imbalance in relation to antioxidant defense mechanisms, which in turn leads to oxidative stress and mitochondrial dysfunction. Mitochondrial dysfunction eventually leads to apoptosis of hepatocytes and thereby, exacerbate the pro-inflammatory events of NASH [17,18]. Besides mitochondria, peroxisomes are also an important site for the generation of ROS through β-oxidation and the activities of numerous oxidases generating H2O2[19]. Peroxisomes not only produce ROS but also serve as a depot of different antioxidative enzymes and molecules [20]. Therefore, any alteration in peroxisomal function can potentially exacerbate the oxidative stress in NASH. In this article, we reviewed the impaired biogenesis and function of peroxisomes in NASH and their role in exacerbating the progression of the disease.
2Why peroxisomes are relevant in NASHPeroxisomes are single membrane-bounded subcellular organelles which are ubiquitous in eukaryotic cells [21]. They have an indispensable role in different metabolic pathways, particularly the breakdown of long-chain fatty acids, very-long-chain fatty acids, and branched-chain fatty acids through a set of α and β-oxidation reactions and metabolism of numerous xenobiotic compounds by respective oxidases and biosynthesis of plasmalogens [21–23]. Throughout these metabolic activities, peroxisomes produce different ROS, such as hydrogen peroxide (H2O2) and superoxide anion (O2−) [24]. In the same time, peroxisomes also contain different antioxidant enzymes and molecules that decompose the ROS produced inside and outside the organelle [25]. Thus, peroxisomes play a double-edged role in the generation and detoxification of ROS [24].
Liver is the key metabolic organ which governs the energy balance of the body [26]. Peroxisomes are most abundant in hepatocytes where they constitute around 1–2% of the total cell volume [27]. Therefore, peroxisomes play a major role in the neutralization of the bulk amount of ROS produced in this metabolically active organ by using ROS-detoxifying enzymes such as catalase and superoxidases. On the other hand, hepatic steatosis leads to an elevated level of free fatty acids within hepatocytes, resulting in an increased production of ROS through the increased fatty acid oxidation [28]. Increased generation of ROS makes the organ more dependent on peroxisomes for the safe disposal of ROS [29]. Therefore, any impairment in peroxisomal function can potentially exacerbate the oxidative stress in the steatotic liver and thereby, facilitate the progression of the disease.
3Impaired peroxisomal function is associated with the development of NASHRegular peroxisomal function in the healthy liver serves two important beneficial roles simultaneously; one is the degradation of very long chain and branched-chain fatty acids, thus preventing them from being accumulated in the liver to induce hepatic steatosis and the other is the scavenging of ROS, and thus preventing oxidative stress [29,30]. To maintain a regular peroxisomal biogenesis and function, it is required to ensure the import of cytosolic matrix protein, formation of peroxisomal membrane and fission of existing peroxisomes [31]. Currently, mutations in 14 PEX genes are known to cause defect in peroxisomal biogenesis [31]. Some of these mutations result in hepatic steatosis, steatohepatitis and fibrosis [32]. For instance, a condition of peroxisomal deficiency created by the liver-specific knockout of Peroxisomal Biogenesis Factor 5 (Pex5) in mice leads to the development of hepatic steatosis [33]. On the other hand, upregulation of peroxisomal biogenesis promotes resistance to dietary fat-mediated hepatic steatosis in mice [34]. These observations provided the initial clue regarding the role of peroxisomes pertaining to the development of hepatic steatosis and NASH.
Peroxisomes contain some enzymes which are exclusive to this organelle and closely associated with the development of NASH, such as acyl-coenzyme A oxidase and catalase. Acyl-coenzyme A oxidase is the rate-limiting enzyme of peroxisomal β-oxidation of long chain fatty acids. Acyl-coenzyme A oxidase null mice show defective peroxisomal β-oxidation, which results in hepatic steatosis and eventually leads to steatohepatitis [35]. Catalase is the H2O2-decomposing enzyme exclusively localized in peroxisomes. Catalase detoxifies H2O2, produced not only in peroxisomes but also in mitochondria, through peroxisome-mitochondria communication [36]. Accordingly, homozygous catalase knock-out results in accelerated oxidative stress and inflammation in the dietary fat-induced steatotic liver in mice [37].
4A key role of PPAR-α in modulating peroxisomal function in NASHPeroxisomal proliferator-activated receptors (PPARs) are nuclear receptor transcription factor family with three isoforms: alpha (α), beta/delta (β/δ), and gamma (γ) [38]. They are differentially expressed in different tissues. While PPAR-β/δ and PPAR-γ are primarily expressed in skeletal muscle and adipose tissue respectively, PPAR-α is most abundantly expressed in the liver [39]. Therefore, PPAR-α is the one mostly relevant to the pathogenesis of NASH. Association of PPAR-α with the transcriptional activation of genes of beta-oxidation is well established [38,40,41]. Therefore, an altered expression of PPAR-α could potentially compromise beta-oxidation while inducing lipogenesis, a major promoting factor for the progression of disease in NASH. Thus, the treatment with PPAR-α-agonist, such as fenofibrate, resulted in upregulated expression of genes involved in hepatic lipid oxidation in high fructose-fed and high fat diet-fed mice [42,43]. Apart from the transcriptional upregulation of genes of beta-oxidation by PPAR-α, Biogenesis and proliferation of peroxisomes are also regulated by this putative transcription factor, in a heterodimeric combination with retinoid X receptor, forming an active multi-protein complex with a variable set of coactivators [44]. Active multi-protein complex binds to peroxisome proliferator response elements located at the promoter regions of most of the genes encoding for peroxisomal proteins [44]. In the livers of NASH patients, expression of PPAR-α decreases and negatively correlates with the presence and severity of the disease [45]. The expression of PPAR-α was also found to simultaneously increase with the improvement of histological NAFLD score secondary to the change of life-style or bariatric surgery [45]. It clearly indicated the hepatoprotective role of PPAR-α in NASH. Park and colleagues had reported that the expression of PPAR-α is reduced in methionine- and choline-deficient diet-induced NASH in mice [46]. Therefore, their study was in line with the aforementioned clinical report. They had also reported that statin-mediated inhibition of hepatic steatosis and NASH recovers the expression level of PPAR-α [46]. Cholesterol is one of the most prominent lipotoxic species that accumulate in the liver during the pathogenesis of NASH. Cholesterol can be exogenously obtained through diet as well as endogenously synthesized from the excess level of acetyl-coA through mevalonate pathway [47]. Statin demonstrates an anti-inflammatory activity through the inhibition of 3-hydroxy-3-methyl glutaryl-coenzyme-A reductase, a rate-limiting enzyme of mevalonate pathway. Inhibition of mevalonate pathway not only results in inhibition of de novo synthesis of cholesterol but also in a deficiency of mevalonate pathway intermediates such as geranylgeranyl pyrophosphate (GGPP) which is required for the prenylation-dependent translocation of RhoA to the cell membrane and activation of RhoA/ROCK pathway [48] (Fig. 1). Because small Rho GTPases and PAPRs exhibit an inverse relationship [49,50], reduced translocation of RhoA to the cell membrane in statin-treated mice eventually results in an increased expression of PPAR-α. Finally, the anti-inflammatory role of PPAR-α in NASH was demonstrated in PPAR-α-null mice as mice deficient in PPAR-α were more susceptible to dietary fat-mediated NASH [51,52]. This association between the downregulated expression of PPAR-α and the development of NASH indicates impaired peroxisomal biogenesis and function in NASH. Due to the anti-inflammatory role of PPAR-α, it is considered as target for the therapeutic treatment of NASH and end-stage liver diseases. For instance, Elafibranor, a dual agonist of PPAR-α/δ, demonstrated efficacy in the treatment of animal models of NASH and fibrosis [53]. It also demonstrated the resolution of NASH without worsening the fibrosis in human patients [54].

Peroxisome-linked pro and anti-inflammatory mechanisms in NASH. 1. Acetyl coA is used to synthesize cholesterol through the mevalonate pathway. Elevated level of cholesterol synthesis contributes to the development of hepatic steatosis and NASH. Inhibition of mevalonate pathway improves hepatic steatosis and NASH. NASH results in increased level of free fatty acids leading to the increased de novo synthesis of cholesterol. 2. cPLA2 and COX2 produce PPAR ligand e.g. eicosanoids. Activated PPAR, in turn, enhances the transcription of catalase and ACOX. Catalase and ACOX contribute to the decomposition of ROS and inhibition of excessive fat accumulation in the liver. Activated ROCK leads to the decreased expression and activation of PPAR. 3. Concentration of circulating LPS is observed to increase in NASH. LPS and endogenous ligands interact with TLR-4, induces NF-κB activation pathway, and enhances the expression of proinflammatory cytokines e.g. TNF-α, which eventually activates the ROCK pathway. NF-κB and PPAR form a negative regulatory loop in NASH. ACOX: acyl-coenzyme A oxidase, AP1: Activator protein 1, COX2: cyclooxygenase-II, cPLA2: calcium-dependent phospholipase A2, NF-κB: nuclear factor kappa B, PPAR: Peroxisomal proliferator-activated receptor, RhoA: Ras homolog gene family member A, ROCK: Rho-associated protein kinase.
In normal condition, hepatocytes have a high expression of PPAR-α [55,56]. Unsaturated fatty acids and their derivatives such as eicosanoids are one of the major natural ligands of PPAR-α which could be produced from phospholipids by calcium-dependent phospholipase A2 (cPLA2) and cyclooxygenase-II (COX2) [57–61]. Therefore, the increased availability of free fatty acids in NASH should increase the biogenesis of peroxisomes through the increased expression of peroxisomal proteins in hepatocytes. But, in reality, it is an opposite scenario. Level of peroxisomal proteins, such as catalase and acyl-coA oxidase, is decreased in NASH [62–64], indicating impaired peroxisomal biogenesis. Impaired peroxisomal biogenesis in NASH may occur at least by two different mechanisms. First, expression of PPAR-α decreases in NASH [45]. It is yet to be clarified why the expression of PPAR-α is decreased in NASH. Expression/activation of Rho-associated protein kinase (ROCK) increases in NASH, which can provide a possible explanation for the reduced expression of PPAR-α in NASH [65]. Lovastatin mediated inhibition of ROCK was shown to induce the expression of PAPRs by Paintlia and colleagues [66]. Therefore, an increased expression/activation of ROCK may have a potential inhibitory effect on the expression and activation of PAPR-α. In addition to ROCK, increased oxidative stress, a common phenomenon in NASH, can also result in altered expression and activation of different transcription factors including PPARs through the regulation of a variety of signaling pathways [67]. Second, peroxisomal number and its biogenesis may also be reduced due to the direct effect of oxidative stress caused by the elevated level of ROS in NASH [18].
Because NASH is known as the hepatic manifestation of metabolic syndrome, likewise NASH, the role of PPAR-α is also well established in insulin resistance and obesity. Treatment of two mouse models of insulin resistance, lipoatrophic A-ZIP/F-1 and MKR mice, with PPAR-α-agonist resulted in decreased insulin resistance [68,69]. On the other hand, combined treatment of rat fed high fat diet with PPAR-α/γ-agonist resulted in complete elimination of triglyceride accumulation in the liver and visceral adiposity [70]. In a number of clinical studies, treatment of type-2 diabetic patients with PPAR-α-agonists showed clear improvement of insulin sensitivity [71]. Although none of these studies looked at the changes in peroxisomal integrity and structure, they commonly indicated a plausible alteration in peroxisomal biogenesis and function in metabolic syndrome.
5Role of TLR-4 pathway in the impairment of peroxisomal function in NASHLiver is the first organ to encounter lipopolysaccharides (LPS) delivered from the gut microbial community through the portal vein [72]. Toll-like receptor-4 (TLR-4) is the major and well characterized pattern recognition receptor of LPS to initiate the LPS-mediated pro-inflammatory events in the fatty liver [73,74]. In liver, TLR-4 is mostly expressed by hepatocytes, monocytes, kupffer cells, and stellate cells [75]. Expression of TLR-4 and downstream mediators is upregulated in the livers of NASH patients compared to the healthy individuals [75]. Observed elevated concentration of serum endotoxin in NASH patients led to investigate the potential role of a sub-clinical dose of LPS in driving chronic inflammation in dietary fat-mediated NASH in experimental models [76–79]. The impact of the TLR-4 pathway in the alteration of peroxisomal proliferation, structure, and function was first indicated in studies by Singh and colleagues [80,81]. They had shown that administration of a sub-lethal dose of endotoxin could induce changes in peroxisomal number, structure, and function in the rat liver [80,81]. Conversely, it was reported that induction of peroxisomal proliferation inhibits the LPS-induced proinflammatory responses in vitro[82]. A possible cross-communication between the TLR-4 pathway and peroxisomal biogenesis was indicated in a study by Necela and colleagues [83]. In this study, they showed that the activation of TLR-4 pathway leads to the downregulation of peroxisomal proliferator-activated receptor-γ (PPAR-γ) via NF-κB-dependent mechanism in macrophages. They also showed that knockout of PPAR-γ had resulted in an increased expression of pro-inflammatory genes [83]. It is also reported that agonist-mediated activation of PPAR-γ suppress the expression of TLR-4 in vascular smooth muscle cells [84]. These observations indicated a negative regulatory loop between TLR-4 pathway and expression of PPAR-γ (Fig. 1). Mechanistically, upregulation of PPAR-γ downregulate the expression of pro-inflammatory cytokines, such as TNF-α, IFN-γ, IL-2, IL-1β, IL-6, MCP-1, and MIP-1β [85]. These cytokines upregulate the transcription of different endogenous ligands of TLR-4, such as high mobility group box-1 (HMGB-1) [86,87]. Thus, increased activation of PPAR-γ results in a decreased activation of TLR-4 pathway. Because PPAR-α and PPAR-γ share commonalities in their ligands and mechanism of inducing gene expression [88], the existence of a similar type of regulatory loop between TLR-4 and PPAR-α cannot be ruled out. Because PPAR-α is the key transcription factor of PPAR family that regulates the expression of genes involved in peroxisomal β-oxidation and proliferation, it is important to investigate the mechanism of a potential crosstalk between TLR-4 and PPAR-α. A better understanding of such communication between TLR-4 and PPAR-α would at least partially explain how peroxisomal dysfunction occurs in NASH. Therapeutic intervention of this TLR-4-PPAR-α loop could improve peroxisomal function and thereby, attenuate the severity of disease in NASH. For instance, TAK-242 is a small compound that selectively inhibits TLR-4 signaling through the interference with interactions between TLR-4 and its adaptor molecules [89]. Therapeutic effect of TAK-242 was shown in mouse models of sepsis and endotoxin-shock [90,91]. It was also trialed for the treatment of severe sepsis in human [92]. Inhibition of TLR-4 pathway by the treatment of TAK-242 could potentially restore peroxisomal function in NASH through the upregulation of PAPR-α.
6ConclusionDue to a major anti-oxidative role in the liver, integrity of peroxisomal biogenesis/function is essential to prevent the oxidative stress-induced inflammation in the fatty liver. A compromised peroxisomal function occurs in NASH which could have a significant contribution to the progression of the disease. As a cause of impaired peroxisomal biogenesis and function, reduced expression of PAPR-α in NASH could be implicated. Nevertheless, it is still not clear why the expression of PAPR-α is decreased in NASH. Investigation on the possibility of formation of a negative feedback loop between the PAPR-α and TLR-4 pathway could explain how the impaired peroxisomal biogenesis and chronic inflammation in NASH are interrelated. Furthermore, such an investigation would also provide an insight into the attenuation of NASH through the therapeutic intervention of peroxisomal biogenetic pathways.AbbreviationsGGPP
geranylgeranyl pyrophosphate
COX2cyclooxygenase-II
cPLA2calcium-dependent phospholipase A2
HMGB-1high mobility group box-1
LPSlipopolysaccharides
NAFLDnonalcoholic fatty liver disease
NASHnonalcoholic steatohepatitis
NF-κBnuclear factor kappa B
PAPR-αperoxisomal proliferator-activated receptor-alpha
PAPR-β/δperoxisomal proliferator-activated receptor-beta/delta
PAPR-γperoxisomal proliferator-activated receptor-gamma
Pex5Peroxisomal Biogenesis Factor 5
RhoARas homolog gene family, member A
ROCKRho-associated protein kinase
ROSreactive oxygen species
TLR-4toll-like receptor-4
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestThe authors have no conflicts of interest to declare.



