




Portopulmonary hypertension is an uncommon but treatable pulmonary vascular consequence of portal hypertension, which can lead to significant morbidity and mortality. Portopulmonary hypertension results from excessive pulmonary vasoconstriction and vascular remodeling that eventually leads to right-heart failure and death if left untreated. Although pulmonary vascular disease in these patients may be asymptomatic or associated with subtle and nonspecific symptoms (dyspnea, fatigue and lower extremity swelling), it should be looked for especially if patients are potential candidates for liver transplantation. Patients with clinical suspicion of portopulmonary hypertension should undergo screening testing, specifically echocardiography. Right heart catheterization remains the gold standard for the diagnosis. The existence of moderate to severe disease poses higher risks and challenges for liver transplantation. The disease has a substantial impact on survival and requires focused pharmacological therapy. New and evolving medical therapies, such as prostanoids (intravenous, inhaled or oral), endothelin receptors antagonists, phosphodiesterases inhibitors, combination therapy and other experimental drugs might change the natural course of the disease. Case reports and cases series have been published regarding the efficacy and safety of pharmacological therapy, but randomized, controlled multicenter trials are urgently needed. Liver transplantation is not the treatment of choice for portopulmonary hypertension, but after optimal hemodynamic and clinical improvement with medical therapy as a bridge, liver transplant can be considered an option in selected patients.
Abbreviations in article:
POPH: Portopulmonary Hypertension
HPS: Hepatopulmonary Syndrome
LT: Liver Transplantation
PAH: Pulmonary Arterial Hypertension
mPAP: mean Pulmonary Arterial Pressure
mPCWP: mean Pulmonary Capillary Wedge Pressure
PVR: Pulmonary Vascular Resistance
TPG: Transpulmonary Gradient
ET-1: Endothelin-1
TTE: Transthoracic Echocardiogram
RVSP: Right Ventricular Systolic Pressure
RHC: Right Heart Catheterization
CO: Cardiac Output
TIPS: Transjugular Intrahepatic Portosystemic Shunt
FDA: Food and Drug Administration
NO: Nitric Oxide
Grants and/or financial support: None
Pulmonary complications have long been documented in the setting of portal hypertension with or without chronic liver disease. Portopulmonary hypertension (POPH) and hepatopulmonary syndrome (HPS) are pulmonary vascular disorders that are associated with portal hypertension and/or liver disease, which can lead to significant morbidity and mortality. Mantz and Craig were the first to describe the association between hepatic and lung vascular disorders in 1951 and since then, a number of case reports and case series have validated this observation.1 As a result of the success of liver transplantation (LT), there has been increasing interest in the diagnosis and management options for these complications of liver disease. Portopulmonary hypertension (POPH) poses difficulties for patients with liver disease and elevated pulmonary artery pressures makes LT more dangerous and in fact may be a contraindication for the LT.2 This review is focused on POPH.
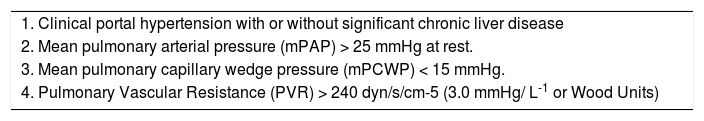
Hemodynamic definitionPOPH is best defined as pulmonary arterial hypertension (PAH) associated with portal hypertension, whether or not portal hypertension is secondary to underlying liver disease.2-5 Current criteria include: a) the presence of portal hypertension (either inferred from the presence of splenomegaly, thrombocytopenia, portosystemic shunts, esophageal varices, or portal veins abnormalities, or may be confirmed by hemodynamic measurements), but not necessarily the presence of cirrhosis; and b) hemodynamic measurements with a mean pulmonary arterial pressure (mPAP) > 25 mmHg at rest, mean pulmonary capillary wedge pressure (mPCWP) < 15 mmHg, and pulmonary vascular resistance (PVR) > 240 dynes/s/cm-5.3 PVR is calculated in the following formula: PVR = [(mPAP – mPCWP)/Cardiac Output] x 80. The criteria applied for the cut-off PVR was agreed upon by a 2004 consensus panel,3 even though some patients with PVR between 120 and 240 dynes/s/cm-5 may be considered to have early or subclinical POPH and should be followed carefully over time.
To properly interpret the hemodynamic changes associated with POPH, it is important to know the most common hemodynamic changes in patients with chronic liver disease. Approximately 30% to 50% of patients with cirrhotic liver disease have low systemic vascular resistance and high cardiac output. In these patients, pulmonary arterial pressures may be elevated due to increased cardiac output, being characteristically important the lower values of PVR.2 These low PVR values that characterize the hyperdynamic state of chronic liver disease has led some investigators to propose that a cutoff of PVR > 120 dynes/s/cm-5 be used in order to define this entity.6 Unfortunately, there are no epidemiological data to support the validity of one definition over the other.
A modified POPH diagnostic criteria is based upon the use of the transpulmonary gradient (TPG) in order to evaluate resistance to flow. TPG = mPAP – mPCWP. A TPG > 12 mmHg is indicative of increased PVR (pulmonary vascular disease), a key characteristic that distinguishes the increase in mPAP associated with POPH (high PVR) from the increase in mPAP due to high cardiac output (low or normal PVR)2,5(See Table I).
Diagnostic criteria for portopulmonary hypertension.
| 1. Clinical portal hypertension with or without significant chronic liver disease |
| 2. Mean pulmonary arterial pressure (mPAP) > 25 mmHg at rest. |
| 3. Mean pulmonary capillary wedge pressure (mPCWP) < 15 mmHg. |
| 4. Pulmonary Vascular Resistance (PVR) > 240 dyn/s/cm-5 (3.0 mmHg/ L-1 or Wood Units) |
The incidence and prevalence of POPH in patients with liver disease is not well defined. In the early 1980’s, an autopsy study of more than 17,000 patients showed pathologic findings of PAH in portal hypertension much more frequently than in others without portal hypertension (0.73% versus 0.13%, respectively).7 More recent studies using the hemodynamic data have estimated the prevalence of POPH in occidental countries to be between 2% to 5%.8,9 In addition, the prevalence in patients undergoing LT is likely higher, with one study showing a prevalence of 8.5%.10
Identification of POPH is made an average of 4 to 7 years after being diagnosed with portal hypertension.11 The mean age of presentation for POPH is in the fifth decade of life, as compared with the third and fourth decade for idiopathic PAH.9
A recent multicenter case-control within a prospective cohort study by Kawut and coworkers,12 examined the clinical risk factors for POPH in patients with advanced liver disease. The sample included 34 cases and 141 controls with normal echocardiographic parameters. Female sex was associated with a higher risk of POPH than male sex (p = 0.018); autoimmune hepatitis was also associated with an increased risk of POPH (p = 0.03), while patients with hepatitis C infection had a lower risk. They concluded that hormonal and immunologic factors may be integral factors for the development of POPH.
Pathobiology and pathophysiologyThe pathogenesis of POPH is not completely understood, but likely involves a complex interaction of several mechanisms. What has been consistently shown in previous studies is that the development of POPH appears to be independent of the cause of the portal hypertension, and the severity of the underlying liver disease does not appear to correlate with the severity of PAH.11 The concept of a vasoproliferative process in PAH has been hypothesized in POPH, causing vascular flow obstruction that leads to increased PVR seen in POPH: an imbalance of vasomediators leading to vasoconstriction, endothelial damage leading to remodeling, with associated proliferation of endothelium and smooth muscle, as well as in situ thrombosis. A proposed mechanism for POPH includes the increased blood flow (high cardiac output) in chronic liver disease causing vascular wall shear stress, which can activate a cascade of events that may eventually lead to the characteristic vasculopathic changes in POPH, and that also could be present in any form of PAH.13 Increased levels of endothelin-1 (ET-1), a potent vasoconstrictor produced by the pulmonary vascular endothelium and the liver, has been shown to play a role in the pathogenesis of idiopathic PAH, as well as in other forms of PAH.14,15 The presence of portosystemic shunts may allow the shunting of the vasoactive substances from the splachnic circulation to the pulmonary circulation, allowing these vasoactive mediators to bypass the liver metabolism and causing substantial effects in the pulmonary vasculature.2-4 Examples of vasoconstrictors include thromboxane-B2 and angiotensin-1, all of which are potent pulmonary vasoconstrictors.16
The development of portosystemic shunts and decrease in the phagocytic capacity of the liver allows circulating bacteria and bacterial endotoxins from the gastrointestinal tract to enter the pulmonary circulation.17 Pulmonary phagocytosis has been demonstrated in cirrhotic patients, suggesting that the induction of interstitial macrophages might contribute to the development of pulmonary vascular disease, such as POPH.17
Both serotonin and ET-1 are dual-action potent pulmonary vasoconstrictive neurohormones that may cause significative vascular remodeling changes and mitogenesis in the pulmonary arteries,18,19 and their abnormal regulation in portal hypertension makes them potentially important candidates in the pathogenesis and pathophysiology of POPH. At the same time, production of vasodilator mediators such as nitric oxide and prostacyclin may be decreased in POPH, facilitating the vascular remodeling and the proliferative vascular response. Importantly, prostacylin synthase as the precursor enzyme for the production of prostacyclin has been demonstrated to be deficient in the pulmonary endothelium of patients with POPH.20
Clinical presentationIn early stages of POPH, patients may be completely asymptomatic or only have symptoms related to their underlying liver disease or portal hypertension. This is very important for clinicians when highly suspecting POPH in their patients, especially in patients that are undergoing evaluation for LT.
Dyspnea on exertion is the most common initial presentation complaint. Other common symptoms include fatigue, generalized weakness, light-headedness, and orthopnea. Physical examination may show elevated jugular venous pressure, an accentuated pulmonic component (P2) of the second heart sound, a murmur that is consistent with either tricuspid regurgitation or a murmur of pulmonic insufficiency (Graham-Steele murmur), as well as a pulsatile liver. Dependent edema out of proportion to ascites may be a sign of right ventricular dysfunction secondary to PAH.21,22 However, dyspnea in a patient with underlying liver disease can stem from a myriad of etiologies, such as cirrhotic cardiomyopathy, renal insufficiency, hepatic hydrothorax, restriction from refractory ascites, parenchymal lung disease or deconditioning and muscle wasting.
Arterial blood gases may show mild to moderate hypoxemia, an increased alveolar-arterial oxygen gradient, and a decreased carbon dioxide levels. A PCO2 < 30 mmHg has been suggested as an indicator of PAH in patients with portal hypertension.23 The decrease in arterial oxygenation was found to be significantly worse in patients with POPH when compared with a cohort of patients that underwent screening for LT and normal right ventricular systolic pressures.24 Electrocardiography may reveal right atrial and ventricular enlargement, right axis deviation, right ventricular strain pattern as well as complete right bundle branch block. Pulmonary function testing may show a decreased diffusing capacity.22 Chest radiography may be normal, but could show enlargement of pulmonary arteries, as well as right-sided cardiac chambers enlargement. Ventilation/perfusion scan may reveal a «mosaic» pattern, but segmental perfusion abnormalities suggests chronic thromboembolic disease.21
Specific evaluation and screeningEvaluation of patients with liver disease or portal hypertension with complaints suggestive of PAH (such as dyspnea on exertion, fatigue and lower extremity edema) will represent the first and most important features in order to suspect the disease. The next and single most important screening test of choice is a two-dimensional transthoracic echocardiogram (TTE).25,26 TTE is pivotal in patients with probable POPH, not only for treatment decisions but for also to assess appropriate candidacy for LT. It is completely unacceptable to make the diagnosis of POPH in the operating room during LT, due to high mortality when those patients have persistent hemodynamic instability during the surgical procedure.27-29
TTE allows the estimation of the right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity using the modified Bernoulli equation, as well as the right atrial pressure, which is fundamental in the initial approach for screening. In the absence of significant pulmonary valve or arterial stenosis, pulmonary arterial systolic pressure equals RVSP. TTE also may show pulmonary insufficiency, right ventricular hypertrophy, dilatation, and dysfunction, and right atrial enlargement in patients who have POPH. Paradoxical septal motion is also common.22 Leftsided valvular abnormalities or left ventricular dysfunction make POPH much less likely. TTE was found to have a sensitivity of 97% and a specificity of 77% in the diagnosis of moderate to severe PAH in patients undergoing screening for LT.28 In another series of patients undergoing LT evaluation, when using an RVSP cutoff of 40mmHg, sensitivity, specificity, positive predictive value, and negative predictive value of TTE were 80%,96%,60%, and 98%, respectively.25
TTE cannot fully discriminate between PAH caused by pulmonary vascular disease (increased PVR) and PAH caused by a hyperdynamic, flow phenomenon state (normal/ low PVR). Therefore, the gold standard for diagnosis of POPH is the right heart catheterization (RHC).30 Recent data from the Mayo Clinic group substantiate advising any patient undergoing LT evaluation with an RVSP > 50 mmHg estimated by TTE to undergo RHC. Even using this cutoff, 34% of patients had normal PVR at time of RHC.29 RHC allows the accurate measurement of the mPAP, PCWP, cardiac output (CO) either by Fick method or thermodilution and calculation of the PVR in order to determine the mechanism causing PAH. Acute vasodilator testing is also usually performed, using either intravenous epoprostenol or inhaled nitric oxide. A decrease in both the mPAP and the PVR of more than 20% with no increased in the CO, can be considered a significant positive response; however this positive response has been mostly studied in the setting of idiopathic PAH.31,32 The aim of this testing in the context of POPH is to aid in determining staging severity (See Table II) and therapeutic expectations, and not to assess for the ability to respond to calcium channel blockers, which is the traditional indication in other forms of PAH.31
Staging of severity of portopulmonary hypertension.
| Variable | Normal | Mild | Moderate | Severe |
|---|---|---|---|---|
| NY HA Class | _ | I, II | II, III | III, IV |
| mPAP (mmHg) | 15-24 | 25-34 | 35-44 | > 45 |
| CI (L/min-1/m2) | 2.5-4 | >2.5 | > 2.5 | < 2.0 |
| PVR (dynes/s/cm-5) | < 240 | 240-500 | 500-800 | > 800 |
| RAP (mmHg) | 0-5 | 0-5 | 5-10 | > 10 |
| Prognosis | - | Favorable | Questionable | Poor |
| Specific Therapy | - | No | Questionable | Yes |
| Reversibility after LT | - | Yes | Questionable | No |
Abbreviations: NYHA = New York Heart Association; mPAP = Mean pulmonary arterial pressure; CI = Cardiac index; PVR = Pulmonary vascular resistance; RAP= Right atrial pressure; OLT = Orthotopic liver transplantation
It is very important and clinically useful to characterize and integrate the pulmonary hemodynamics that complicate liver disease into 3 following subsets on the basis of measured hemodynamic outcomes, such as mPAP, mPCWP, CO and calculated PVR in the stable resting state. 1) The hyperdynamic state will show a minimal increase in mPAP with increased CO, due to passive distention of compliant arteries and recruitment of upper lung arteries.9,33 This subgroup is the most frequent finding in liver disease, including HPS patients. 2) Increased pulmonary venous volume. Volume increase reflects probable excess of volume and/or pressure increase due to limitation in pulmonary blood flow to the left atrium because of left ventricular dysfunction (systolic or diastolic). This results in an increase of mPCWP. This subset is very common in advanced alcoholic-related cirrhosis patients.33 3) Fixed pulmonary vascular disease due to obliterative changes in the vasculature, true POPH. Here the pathological features of POPH include persistent vasoconstriction as well as endothelial and smooth muscle proliferation, fibrosis as well as thrombosis in situ.34
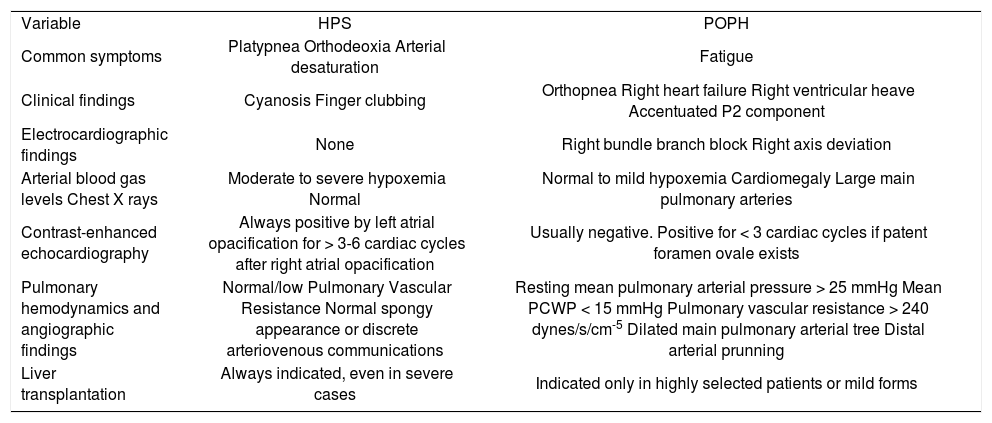
The following table shows the principal clinical and diagnostic features between POPH and HPS. (See Table III).
Clinical, diagnostic and therapeutic distinctions between hepatopulmonary syndrome (HPS) and portopulmonary hypertension (POPH).
| Variable | HPS | POPH |
| Common symptoms | Platypnea Orthodeoxia Arterial desaturation | Fatigue |
| Clinical findings | Cyanosis Finger clubbing | Orthopnea Right heart failure Right ventricular heave Accentuated P2 component |
| Electrocardiographic findings | None | Right bundle branch block Right axis deviation |
| Arterial blood gas levels Chest X rays | Moderate to severe hypoxemia Normal | Normal to mild hypoxemia Cardiomegaly Large main pulmonary arteries |
| Contrast-enhanced echocardiography | Always positive by left atrial opacification for > 3-6 cardiac cycles after right atrial opacification | Usually negative. Positive for < 3 cardiac cycles if patent foramen ovale exists |
| Pulmonary hemodynamics and angiographic findings | Normal/low Pulmonary Vascular Resistance Normal spongy appearance or discrete arteriovenous communications | Resting mean pulmonary arterial pressure > 25 mmHg Mean PCWP < 15 mmHg Pulmonary vascular resistance > 240 dynes/s/cm-5 Dilated main pulmonary arterial tree Distal arterial prunning |
| Liver transplantation | Always indicated, even in severe cases | Indicated only in highly selected patients or mild forms |
The pharmacological management of POPH has been based largely on data from the treatment of other forms of PAH. Treatment of mild POPH (mPAP < 35 mmHg) is debatable. In most of these cases many patients are not even symptomatic, but on the other hand more symptomatic from their chronic liver disease, and they have good functional status. As a group, such patients have not been formally included in original investigations to date.35 Likely secondary to the rarity and also exclusion of patients with POPH, treatment of this entity has been almost empirical, lacking randomized, controlled clinical trials. The usual approach in treating PAH from any cause has been to initiate therapy when the patient is symptomatic and his or her resting mPAP > 25 mmHg as well as elevated PVR.36,37
General therapyThis includes diuretics, which function by reducing the increased intravascular volume commonly present in chronic liver disease. In addition, diuretics can reduce the CO by decreasing the right ventricular preload. Furosemide and spironolactone are the traditional diuretics of choice.25
Mild to moderate arterial hypoxemia is a common finding in POPH, as previously discussed. Theoretically, hypoxemia may worsen PAH through pulmonary vasoconstriction, and therefore supplemental oxygen should be considered for all patients with hypoxemia to maintain oxygen saturation higher than 90% at all times.2,3 Assessment of nocturnal hypoxemia (obstructive sleep apnea) should be accomplished and treated when abnormal.
Transjugular intrahepatic portosystemic shunt (TIPS) has no role in the management of POPH, and may actually be deleterious due to increase in the preload, thereby increasing CO and mPAP. Such acute changes in hemodynamics may place additional stress on an already dysfunctional right ventricle in the setting of moderate to severe POPH.23
Anticoagulation, which is often given for other forms of PAH for its ability to slow disease progression, is traditionally not recommended in patients with POPH.3 This is because of the inherent risk of hemorrhagic complications in patients with underlying liver disease and portal hypertension, especially in patients with a prior history of gastrointestinal bleeding.
Vasodilators and novel therapiesCalcium channel blockers, which are often first-line vasodilators in patients who have a positive response to the acute vasodilator challenging test during RHC, are contraindicated in POPH, because it has been demonstrated that they could increase the hepatic venous pressure gradient.38,39 They could exacerbate mesenteric vasodilatation that, in turn, results in higher flow into the portal circulation, increasing the portal vascular pressure, thus worsening the pressure difference between the portal and the hepatic veins.
Epoprostenol is an intravenous analogue of prostaglandin I2, acting as a pulmonary and systemic vasodilator, as well as an inhibitor of platelet aggregation. It has a very short half life, requiring mixing, and must be kept cold with ice packs. Epoprostenol is not an easy treatment to administer due to it’s continuous intravenous delivery system that requires permanent central venous access, and the risk of complications. Common adverse effects attributable to this drug include jaw pain, headache, diarrhea, flushing, leg pain and vomiting.40 The interruption of the infusion may be life-threatening because of the sudden loss of vasodilatation and rebound vasoconstriction.
This drug has been the best studied in POPH. In the largest series to date, epoprostenol was shown to improve the mPAP and the PVR in both the acute and long-term settings.41 Smaller series have shown similar results;42-44 however, whether this improvement is translated to an improvement in survival is unclear. Preliminary data indicate that epoprostenol plus LT in highly selected patients does improve survival.45 An important concern with epoprostenol are the reports of progressive splenomegaly with worsening thrombocytopenia;46 however, recent data from Mayo Clinic does not demonstrate a statistically significant decrease in platelet count in a large cohort of patients with POPH treated with epoprostenol.47 Other prostacyclin analogues (treprostinil and iloprost) have been tested in case reports and case series in POPH. Long-term subcutaneous treprostinil infusion resulted in an improved six-minute walk test.48 A single patient was tested acutely with intravenous iloprost, resulting in a 26% decrease in mPAP and 42% fall in PVR49 over a 12 month-period of follow-up. Another patient with severe POPH was successfully bridged to OLT with continuous intravenous iloprost, the patient’s clinical course > 2 years after LT was excellent, without the recurrence of POPH.50
In portal hypertension, increased production and release of ET-1, a potent vasoconstrictor and smooth muscle mitogen, occurs in the liver,14 the intestines, and the spleen.51 ET-1 levels have been also elevated in patients with decompensated liver cirrhosis and POPH as compared as those with decompensated cirrhosis without POPH.14 For this reason, the use of an oral endothelin receptor antagonist might be an adequate option for therapy. The dual endothelin receptor antagonist, bosentan, is an oral agent approved by the Food and Drug Administration (FDA) in 2002 for treatment of PAH.21,32,52 This agent may cause a transient increase in the aminotransferases, of significant concern in the POPH population. The mechanism of action of this medication is the nonselective inhibition of the endothelin-1, subtypes A and B on the endothelial and vascular smooth muscle cells.53 In approximately 10% of patients with PAH, bosentan can cause elevations of aminotransferase, alkaline phosphatase, and bilirrubin levels. Therefore it is recommended to monitor them on a monthly basis.54
So far, case reports and a retrospective case series of patients with POPH in Child’s class A have showed clinical, functional, and hemodynamic benefits without significant elevation of hepatic aminotransferases on treatment with bosentan.55-58 Hoeper and coworkers,57 studied 11 consecutive patients with cirrhosis and POPH in New York Heart Association functional classes III and IV that were treated with bosentan for > 1 year. After 1 year of therapy, all patients showed improvement of symptomatology and exercise capacity, an increase in the sixminute walk test (p < 0.004), as well as hemodynamic improvement reflected in a statistically significant decrease in the PVR (p = 0.007). Bosentan was well tolerated by all patients, and there was no evidence of drug-related liver injury. This small series of patients, bosentan proved to be efficacious and safe in patients with POPH.
Hoeper and coworkers published a subsequent retrospective analysis which assessed the safety and efficacy of inhaled iloprost, a prostacyclin analogue, and bosentan in additional patients with POPH.59 Thirty-one patients with Child A or B class liver cirrhosis and severe POPH were treated for up to three years with either iloprost (n = 13) or bosentan (n = 18) and the effects on exercise capacity, hemodynamics and survival were evaluated. Event-free survival rates, such as survival without OLT, severe right heart failure or clinical worsening (p = 0.017), requiring the initiation of a novel therapy for PAH, was significantly better in the bosentan cohort study group. Bosentan also had significantly better effects than inhaled iloprost on exercise capacity, determined by the six-minute walk test, as well as on hemodynamics. In this series, therapy with both inhaled iloprost and bosentan appeared to have a positive response and was safe, but with higher survival rates in the bosentan group at 3 years when compared to the iloprost group (94, 89 and 89% vs 77, 62 and 46%, respectively). This retrospective analysis had some limitations that included the small number of patients, was not randomized, treatments were not blinded and the impact of the treatment on portal hypertension was not investigated. This will be an important issue for future studies, especially since two case reports have described reduction of portal venous pressure with bosentan therapy.58,59
Ambrisentan, is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of PAH.60 It has not yet been studied in patients with POPH, however, as in previous studies, elevations in liver enzymes and bilirrubin may also occur, and monthly monitoring is indicated. Preliminary data from the Mayo Clinic suggest this drug may significantly decrease PVR and mPAP after 12 months of therapy with no adverse effects on liver function (Michael J. Krowka, MD, personal communication).
Another oral agent that might be effective in POPH is the phosphodiesterase-5 inhibitor sildenafil, which was approved by the FDA in 2005 for the treatment of PAH.61,62 Sildenafil blocks the degradation of cyclic guanosine monophosphate, the second messenger of nitric oxide (NO), thereby prolonging the NO-mediated vasodilatation. In other forms of PAH, sildenafil increases CO, and decreases both the mPAP and PVR, without major adverse effects.61
In one case report, therapy with sildenafil, in a patient with severe POPH (mPAP > 58 mmHg), the mPAP decreased down to 28-30 mmHg, and the patient underwent successful liver transplantation.5,63 Reichenberger and coworkers64 published an uncontrolled study in 14 patients with moderate to severe POPH from diverse etiologies with oral sildenafil. Eight patients were newly started on sildenafil, while 6 patients were already on therapy with inhaled prostanoids (iloprost in five and treprostinil in one). Sildenafil appeared to provide therapeutic benefit (decreasing the mPAP and PVR) when evaluated at 3 months, but the hemodynamic benefit was not sustained after 12 months in 7 patients, as measured by PVR, mPAP and CO. Curiously, the six-minute walk test continued to improve at 3 and 12 months (p < 0.01 and p < 0.00001 from baseline at three and twelve months, respectively). In an editorial presented by Krowka and coworkers,65 they suggest that the vasodilatation approach provided by sildenafil alone may not be the optimal treatment in moderate to severe POPH, with the option being to offer combination, synergistic therapies with a pulmonary vasodilator and antiproliferative mechanisms as a reasonable approach.
Beta-blockers have been used in many patients with liver disease as both primary and secondary prophylaxis for variceal gastrointestinal bleeding. Beta-blockers prescribed for this indication are assumed to contribute to deterioration of PAH because of their negative inotropic and chronotropic effects.3 Provencher and coworkers recently demonstrated that in the context of moderate to severe POPH, withdrawing beta-blockers improved exercise capacity and pulmonary hemodynamics, concluding that beta-blockers are contraindicated in patients with moderate to severe disease.66 Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn. Beta-blocker therapy in POPH requires special considerations and if at all possible.5
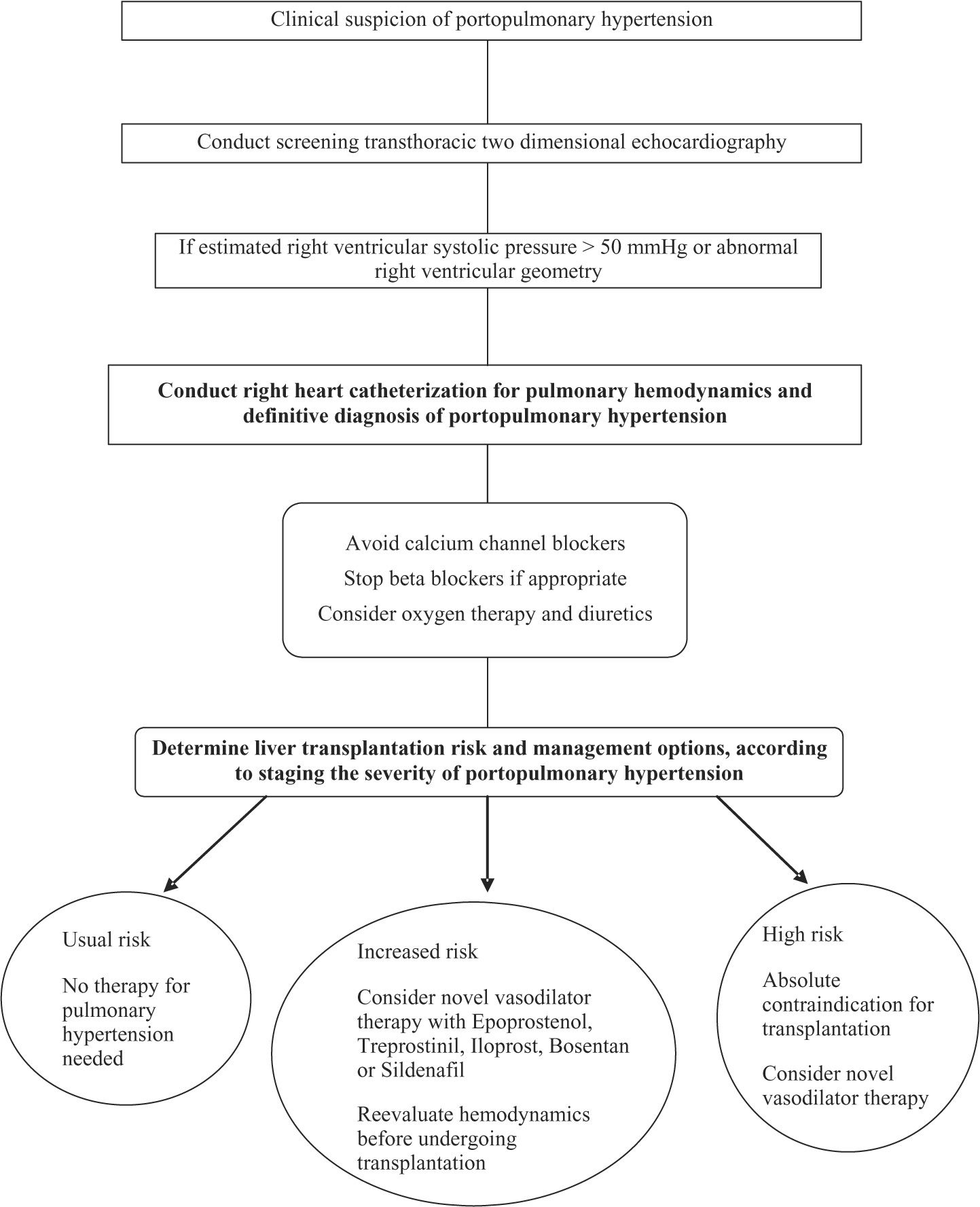
Liver transplantationLT may be beneficial in only highly carefully selected patients with POPH. The presence of POPH increases the risk for perioperative, as well as long-term, morbidity and mortality associated with LT.67 As mPAP and PVR increase, so does the mortality associated with LT.68 Many, if not all, LT centers consider mPAP higher than 50 mmHg to be an absolute contraindication to LT.2-5 Potential candidates must be evaluated at experienced centers in the integrative management of POPH. Clinical assessment protocols before LT must include an algorithmic approach. One proposed algorithm for patients undergoing LT evaluation uses screening TTE, followed by RHC, if the estimated RVSP is > 50 mmHg, and then staging patients according to the POPH severity in order to guide appropriate management (See Figure 1).

When patients without POPH undergo LT, right ventricular function is preserved throughout all phases of the surgery (68).In patients with POPH, however, they may develop hemodynamic instability during LT. The most critical times are the induction of the anesthesia, during and after the graft reperfusion, and the immediate postoperative period.69,70
For LT candidates with documented moderate to severe POPH (mPAP > 35 mmHg) who will have vasodilator therapy initiated, the goal of therapy should be to reduce the mPAP to < 35 mmHg and the PVR < 400 dynes/ s/cm-5 before proceeding to LT.3 The reason for this cutoff is that previous data have demonstrated no increased mortality risk when the mPAP is < 35 mmHg.9,27 A recent report from Sussman and coworkers,71 demonstrated that in 8 patients with POPH as their only contraindication for LT, pretreatment with intravenous epoprostenol caused a significant improvement in their hemodynamics, allowing 75% to be listed for LT. There have been reports of persistence of progression of PAH even after LT.72,73 Recurrence of PAH following failure of the transplanted liver can also occur.74
A multicenter database was organized at 10 LT referral centers to identify the outcome of patients with POPH being evaluated for LT between 1996 and 2001.67 Interestingly the database showed that the in-hospital LT mortality was 36% in POPH patients, reemphasizing the need for accurate preoperative diagnosis and assessment. Death in these patients resulted from right-sided heart failure.23,67
Prognosis and survivalSurvival from time to diagnosis is difficult to predict, and the natural history of untreated POPH varies with the degree of liver disease and the severity of PAH. Transplant-free survival was 85% at one year and 38% at three years in one study performed by Kawut and coworkers.75 In that study they demonstrated a higher risk of mortality in patients with POPH as compared with patients with idiopathic PAH, despite having better hemodynamic measurements in the POPH group.
In a retrospective analysis from Robalino and coworkers in 78 patients treated conservatively (before the era of prostanoids or oral drugs became available), the median survival was 6 months from the time of diagnosis.76
In a recent retrospective screening right-heart catheterization survival analysis by Swanson and colleagues77 from the Mayo Clinic Liver Transplantation Group, identifying 74 POPH patients from 1994 through 2007, and categorizing patients in to 3 groups: 1) no therapy for POPH or LT, 2) therapy for POPH alone and 3) therapy for POPH followed by LT. Nineteen patients received no therapy for POPH and no LT representing the natural history of POPH. Five-year survival was 14%, with 54% dying within 1 year of diagnosis. Fortythree of the 74 patients (58%) received medical therapy for POPH and did not undergo LT. The median survival was 46 months and 5-year survival was 45%, being significantly better than the patients who were not selected to receive medical therapy for POPH (p = 0.03). Twelve patients underwent LT and 5-year survival for the 9 patients receiving therapy for POPH was 67% as compared as 25% in 3 patients who were not pretreated with prostacyclin therapy. Interestingly, there were no differences in the pulmonary hemodynamics (mPAP, PVR or TPG) or severity of liver disease between survivors and non-survivors suggesting that in POPH we cannot predict who is going to do poorly based on pulmonary hemodynamics or liver disease severity alone. In conclusion, the survival of untreated patients with POPH was very poor, with the subgroup of patients selected to medical therapy with or without LT having better long-term survival. Medical therapy with pulmonary novel vasomodulators was advised in all patients with significant POPH.
Unfortunately most of the data on outcomes of pharmacological therapy and LT come from case series and retrospective analysis; prospective trials have been lacking. However, some patients with moderate to severe POPH have been bridged with medications to lower mPAP and PVR so that LT could be safely done, and some have been able to discontinue pharmacotherapy due to resolution of their PAH after LT.41,63,71
Conclusion and future directionsPOPH could be considered now one of the uncommon complications of portal hypertension and liver disease, focusing the attention in LT referral centers to assess and properly approach this particular population. The recent advances in the understanding of the pathobiology and pathophysiology in PAH make POPH a potential target for the novel therapeutic armamentarium that has been developing in the last decade in other forms of PAH.
Lacking animal models of POPH makes difficult further advances in the pathogenesis, diagnosis and therapy of this disease. Researchers are now attempting to establish animal models. POPH management requires the cooperation of wide knowledgeable physicians of multidisciplinary specialties in order to have better care and outcomes, including the pulmonologist/intensivist, hepatologist, cardiologist and transplant surgeon.
Appropriate randomized multicenter clinical trials are going to be essential to establish more solid evidencebased guidelines for the pharmacological management of POPH, especially in the potential candidates for OLT after achieving the appropriate clinical, functional, and hemodynamics goals. One way to gain more knowledge regarding the response to novel therapies in POPH patients could be to include this population in future randomized, clinical trials, that have as a primary end-point the efficacy and safety of new molecules, such as oral endothelin receptor antagonists, phosphodiesterase inhibitors, intravenous and inhaled prostanoids or even combined synergistic therapy.76 Also, recent animal and human experimental pharmacological studies with new agents (e.g. serotonin transport inhibitors, statin therapy, and platelet-derived growth factor inhibitors) in POPH are now being evaluated in phase one trials and sound attractive for the clinical researcher.
Documenting a relationship between genetic polymorphisms in POPH has potential therapeutic implications. If such abnormalities can be detected in patients with advanced liver disease in the early hyperdynamic phase (low/normal PVR and normal TPG), innovative measures might be undertaken to prevent the evolution into full-blown complex arteriopathic proliferative/obliterative process of this devastating disease.



