





The hepatoprotective potential DTS (1.5 g/kg bw, Densh-ici-to-Chiusei, Kyotsu Jigyo, Tokyo, Japan) was evaluated against either toxic (1.5 g/kg bw) and sub-toxic (150 mg/kg bw) dosage of paracetamol-induced liver injury in Sprague-Dawley rat. Paracetamol intoxication caused a reduction of serum total protein and increase levels of serum alkaline phosphatase (ALP), aspartate ami-notranferase (AST) and serum alanine aminotranferase (ALT) at higher extent in the toxic group. This phenomenon was paralleled by an impaired liver redox status (reduced glutathione (GSH), superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT) and increased MDA in both paracetamol-administered groups. Moreover, a marked reduction of AT-Pase and thiols together with DNA fragmentation occurred in liver tissue. Animals pretreated with DTS showed a marked mitigation of the severity of liver enzyme and of the impaired redox status of the liver. Moreover, DTS partly prevented the DNA fragmentation and the decline of liver tissue ATPase and protein thiol assay as compared with both groups treated with paracetamol alone. Although more detailed studies are awaited to ascertain the detailed mode of action of DTS, it wouls seem to be related to the prevention of formation of the reactive oxygen groups thereby preventing the damage on the hepatocytes and possibly modulating the genes responsible for synthesis of liver antioxidant enzymes thus providing marked DNA protection.
Drug-induced liver injury is a potential complication of virtually every prescribed medication, because the liver occupies a central role in the metabolic disposition of all drugs and foreign substances. Most of the hepatotoxic chemicals damage liver cells mainly by lipid peroxida-tion and other oxidative damages and this applies also to paracetamol which is a widely used analgesic/antipyretic agent regarded as generally safe when used at therapeutic levels1 while representing the drug of choise in children. However, paracetamol hepatotoxicity is the leading cause of drug-induced liver failure in the western countries2 and an acute or cumulative overdose can cause severe liver injury with the potential to progress to liver failure.
The main toxicity mechanism advocated for include the Cyp2E1 metabolic activation of the reactive metabolite, N-acetyl-p-benzoquinone imine which depletes cellular glutathione and then covalently binds to critical cellular proteins and macromolecules.3,4
The following alkylation of proteins, namely mito-chondrial proteins,5 on its turn, triggers the formation of reactive oxygen species into the mitochondria.6,7 These events are primarily based on the dysfunction of the cellular Ca2+ homeostasis, with enhancement of the cytosol-ic Ca2+ concentration, noxious translocation of Bax and Bid to the mitochondria and peroxynitrite formation too.
Superoxide anions insofar generated can dismutate to form molecular oxygen and hydrogen peroxide, which then require electrons from GSH molecules to be reduced to water by glutathione peroxidase enzyme and brings about a significant increase of mitochondrial glutathione disulfide (GSSG) levels.6,7 Indeed, while Heinloth et al.8 has reported that a sub-toxic (150 mg/kg) dose of paracetamol would not produce substantial histologically-de-tectable damage in the liver of experimental rats, more recently, it has been shown that even in such event a remarkable 30% depletion of GSH content takes place together with a significant accumulation of 8-OH-dG DNA and nitro-tyrosine protein adducts in the liver.
Therefore, although lipid peroxidation does not seem to be the main cause of cell injury,9 the oxidant stress remains a critical phenomenon in the disruption of mito-chondrial membrane permeability transition pores and the derangement of membrane potential,10,11 with ATP depletion and cell death due to oncotic necrosis.12,13 This may hamper, to a different extent of severity, the major functions of the liver such as detoxification of bilirubin, epimerization of galactose to glucose as uridine-5-phos-phate derivatives, synthesis of protein and prothrombin, handling of enzymes, such as alkaline phosphatase (ALP), release of aspartate aminotranferase (AST) and ala-nine aminotranferase (ALT).
Moreover, it has also been shown, both in vivo and in cultured hepatocytes, that, in parallel to liver damage paracetamol overdose may also determine a recognizable fragmentation of nuclear DNA and karyolysis.14-16 GSH, one of the major tripeptide non-enzymatic biological an-tioxidants present in the liver, is committed with the removal of free radicals and maintenance of membrane protein and thiols, and a substrate for GPx.
We have recently shown that DTS, a novel nutraceuti-cal, might exert a beneficial regulation of GSH/GSSG re-dox status while increasing also glutathione reductase activity and mitochondrial SOD fraction.17,18 Thus, in view to an integrated approach aimed to find a possible protective natural counteraction against drug-induced side effects, in the present study we investigated if DTS would play a significant hepatoprotective action in rats whose with high-dose paracetamol.
Materials and methodsAnimalsAdult Sprague-Dawley rats of either sex (150-180 g) were used throughout the experiments and were maintained under controlled standard conditions of light (12/ 24 h) and temperature (26 + 1 °C). Food pellets and tap water were provided ad libitum. For experimental purposes animals were fasted overnight but were allowed free access to water. All animal procedures were performed according to approved protocols and in accordance with the recommendations for the proper care and use of laboratory animals.
Preparation of phytotherapeutic compound. DTS (panax pseudoginseng, eucommia ulmoides, ginseng radix, in the weight percentage of 50%/25%/25%, Kyotsu Jigyo, Tokyo, Japan) is produced under quality-controlled proceduresin non OGM-modified crops and ISO 9001 and 140001 regulation, registered as FOSHU (Food Of Specific Health Use) and it was kindly donated by the Institute of Health Care with Oriental Herbs and Medicine, Tokyo, Japan. This compound presents in the form of tiny grains of medium consistency and palatable which can be easily mixed with food.
Study design. The rats were divided into 5 groups comprising 15 animals in each group. Group A was maintained as the control. Group B (acute model) received paracetamol (Sigma Chemical Co., USA) suspension (1.5 g/kg bw) as a single dose. Group B-tx received paracetamol (1.5 g/kg bw) followed by DTS (150 mg/kg). Group C (sub-toxic model) given paracetamol (150mg/kg bw) as a single dose orally and group C-tx, same dosage of paracetamol plus DTS (150 mg/kg). Sacrifice were carried out 24 hours after «acute» and «sub-toxic» dosage.
Biochemical assessment. At the end of the experimental period, animals were fasted for 12 h and blood samples were obtained from the experimental and control rats by puncturing retro-orbital plexus. Aspartate aminotrans-ferase (AST) and alanine aminotransferase (ALT) activities in serum were measured with a spectrophotometric method, whereas colorimetric determination of alkaline phosphatase (ALP) activity was carried using commercial kits. After collection of blood samples the rats were sacrificed by cervical dislocation, and the livers were immediately excised, rinsed in ice cold normal saline followed by 0.15 mol/L Tris-HCl, dried, and weighed. Glutathione peroxidase (GPx) activity was assayed at 340 nm by spectrophotometry and the amount of the enzyme converting 1μmol GSH per min was taken as 1 activity unit. Glutathione reductase activity was measured at 340 nm by spectrophotometry and the amount of the enzyme reducing 1μmol GSSG per min was regarded 1 activity unit as elsewhere described.19 Superoxide dismutase activity (SOD) was measured at 560 nm as the rate of suppression of reduction of nitrotetrazolium blue and for 1 unit of activity, the amount of protein was taken which provided 50% inhibition of nitrotetrazolium blue reduction under standard conditions.20 Catalase (CAT) activity was calculated at 240 nm by measuring the rate of H2O2 utilization with the molar extinction coefficient for H2O2 being 43.6 M-1 cm-1. The amount of the enzyme utilizing 1μmol H2O2 per min was taken as 1 activity unit.21 Malondyalde-hyde (MDA) determination. MDA in liver was assayed by spectrophotometric method and the concentration of thiobarbituric acid was calculated by the absorbance coefficient of MDA-TBA complex 1.56 x 105 cm-1 M-1 and expressed in nmol/mL.22 The amount of phosphorus released by the enzymes was calculated as a quantitative measure of the activities of total ATPase.23 Tissue protein thiols were determined according to the methods described by Sedlack and Lindsay24 by using Ellman’s re-agent (5,5’-dithiobis 2-nitrobenzoic acid; DTNB), which was reduced by thiol groups to form 1 mol 2-nitro 5-mer-captobenzoic acid/mol thiol and with maximal absorption at 412 nm.
Evaluation of DNA fragmentation. DNA fragmentation was measured by the diphenylamine (DPA) spectro-photometric method.25 Intact DNA was separated from frag-mented DNA by centrifugal sedimentation followed by precipitation and quantification using DPA. To minimize formation of oxidative artifacts during isolation, 2,2,6,6-tetramethylpiperidinoxyl (20 mM final concentration) was added to all solutions and all procedures were performed on ice. Briefly, hepatocytes (1 x 106 in 1 mL PBS) were put in a 1.5 mL centrifuge tube (tube B) and centrifuged (200 g, 4°C, 10 min) to obtain a cell pellet. The supernatants were transferred to fresh tubes (tube S). The obtained pellet (tube B) was suspended in 1 mL TTE (Tris Triton EDTA) buffer, pH 7.4 (TE buffer with 0.2% Triton X-100) and centrifuged at 20,000 g (4 °C, 10 min). The supernatant obtained was transferred to fresh tubes (tube T) and the resulting pellets were resuspended in TTE buff-er. TCA (tri-chloro acetic acid, 1 mL of 25%) was added to tubes T, B and S and vortexed vigorously. Tubes were kept overnight at 4 °C followed by centrifu-gation at 20,000 g (4 °C, 10 minutes). The supernatant was discarded and the pellet was hydrolysed by the addition of 160 pl of 5% TCA fol-lowed by heating at 90 °C for 15 minutes. This was followed by addition of 320 μL of freshly prepared DPA. The colour was developed by incubation at 37 °C for 4 hours. Optical density of the solution was read at 600 nm in an ELISA reader. Percentage DNA Fragmentation was calculated using the following formula: % Fragmented DNA = S + T/ S + T + B x 100. An agarose gel electrophoresis was also performed to analyse DNA fragmentation as described elsewhere.22
Statistical analysisData was analyzed by ANOVA using Duncan’s post-hoc test for comparisons among means at p • •0.05 when appropriate. If the data were not normally distributed, we used the Kruskal-Wallis test (nonparametric analysis of variance) followed by Dunn’s multiple comparisons test. Statistically, p < 0.05 was considered significant.
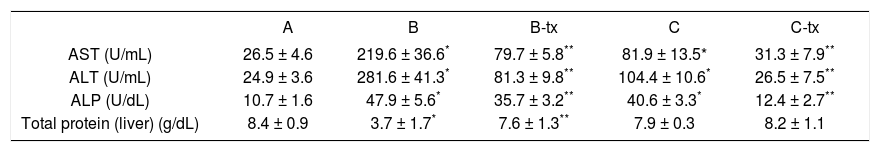
ResultsSerum and liver tissue biochemistry. The extent of paracetamol induced hepatotoxic effect was assessed by the levels of released cytoplasmic enzymes such as ALP,
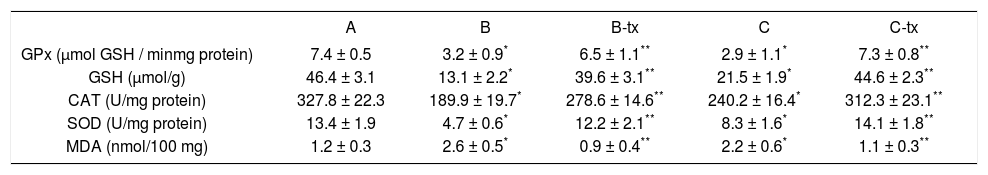
AST and ALT in circulation. The levels of serum tran-saminases and total protein in the normal, paracetamol injured and DTS-administered rats are shown in table I. The levels of AST, ALT and ALP increased significantly in the paracetamol-treated rats, while the content of protein decreased significantly when compared to the control. These features were more pronounced in toxic dosage versus sub-toxic group (p < 0.01). Treatment with DTS was found to reduce the concentration of AST, ALT and ALP while increasing the protein content in B-tx (p < 0.01) while normalizing such parameters in C-tx (p < 0.05). Table II shows the effects of administration of DTS on the levels of liver MDA, GSH, SOD, CAT and GPx in the toxic and sub-toxic paracetamol-treated rats. Both paracetamol dosages brought about a significant depletion of GSH, SOD, CAT and GPx with increased MDA (p < 0.01; B vs C: p < 0.05). The level of non-enzymatic and enzymatic antioxidants in both DTS-treated groups were found to be higher than the untreated rats with significantly lower MDA generation (p < 0.05). In particular, DTS administration brought about a full restoration of all the above redox parameters in C-tx group (p < 0.001).
Plasma level of liver enzyme after paracetamol loading with toxic (B: 1.5 g/kg) and sub-toxic (C: 150 mg/kg) dose and DTS co-treatment (TX).
| A | B | B-tx | C | C-tx | |
|---|---|---|---|---|---|
| AST (U/mL) | 26.5 ± 4.6 | 219.6 ± 36.6* | 79.7 ± 5.8** | 81.9 ± 13.5* | 31.3 ± 7.9** |
| ALT (U/mL) | 24.9 ± 3.6 | 281.6 ± 41.3* | 81.3 ± 9.8** | 104.4 ± 10.6* | 26.5 ± 7.5** |
| ALP (U/dL) | 10.7 ± 1.6 | 47.9 ± 5.6* | 35.7 ± 3.2** | 40.6 ± 3.3* | 12.4 ± 2.7** |
| Total protein (liver) (g/dL) | 8.4 ± 0.9 | 3.7 ± 1.7* | 7.6 ± 1.3** | 7.9 ± 0.3 | 8.2 ± 1.1 |
Redox status in the liver after paracetamol loading with toxic (B: 1.5 g/kg) and sub-toxic (C: 150 mg/kg) dose and DTS co-treatment (TX).
| A | B | B-tx | C | C-tx | |
|---|---|---|---|---|---|
| GPx (μmol GSH / minmg protein) | 7.4 ± 0.5 | 3.2 ± 0.9* | 6.5 ± 1.1** | 2.9 ± 1.1* | 7.3 ± 0.8** |
| GSH (μmol/g) | 46.4 ± 3.1 | 13.1 ± 2.2* | 39.6 ± 3.1** | 21.5 ± 1.9* | 44.6 ± 2.3** |
| CAT (U/mg protein) | 327.8 ± 22.3 | 189.9 ± 19.7* | 278.6 ± 14.6** | 240.2 ± 16.4* | 312.3 ± 23.1** |
| SOD (U/mg protein) | 13.4 ± 1.9 | 4.7 ± 0.6* | 12.2 ± 2.1** | 8.3 ± 1.6* | 14.1 ± 1.8** |
| MDA (nmol/100 mg) | 1.2 ± 0.3 | 2.6 ± 0.5* | 0.9 ± 0.4** | 2.2 ± 0.6* | 1.1 ± 0.3** |
Group A: control: B: «acute toxic» model; B-tx «acute toxic» model plus DTS 150 mg/kg; C: «sub-toxic» model; C-tx: «sub-toxic» model plus DTS 150 mg/kg. GPx: glutathione peroxidase; GSH: reduced glutathione; CAT: catalase; SOD: superoxide dismutase; MDA: malonyldialdehyde
p < 0.01 vs control values
Liver tissue ATPase and protein thiol. The level of hepatic tissue total ATPase and of tissue protein thiol in control and experimental rats are shown in Figure 1. Group B and C animals intoxicated with paracetamol showed a significant decrease in levels of total ATPase compared with healthy control animals (p < 0.01). Group B-tx and C-Tx animals showed a significant prevention in the impairment of ATPase activity and of tissue protein thiol as compared with their paracetamol intoxicated counterpart. Animals intoxicated with either dosage of paracetamol showed also an increased tissue level of calcium as compared to healthy control (μg/mg protein: B: 18.2 ± 0.9; C: 16.2 ± 0.6 vs A: 10.2 ± 0.4 p < 0.01, data not shown). This abnormality was prevented in both DTS-treated animals (p < 0.05).
; B-tx «acute toxic» model plus DTS 150 mg/kg; C: «sub-toxic» model (150 mg/kg); C-tx: «sub-toxic» model plus DTS 150 mg/kg. *p < 0.01 vs control values **p < 0.05 vs baseline values")
Total ATPase and protein thiols during toxic and sub-toxic paracetamol administration: effect of DTS.
Group A: control: B: «acute toxic» model (1.5 g/kg); B-tx «acute toxic» model plus DTS 150 mg/kg; C: «sub-toxic» model (150 mg/kg); C-tx: «sub-toxic» model plus DTS 150 mg/kg.
*p < 0.01 vs control values
**p < 0.05 vs baseline values
Evaluation of DNA fragmentation. The severity of DNA fragmentation in B and C groups animals administered either dosage of paracetamol was remarkably increased and at a similar extent (p < 0.005 vs control, Figure 2). Groups B-tx and C-tx animals pretreated with DTS showed considerable mitigation of acute DNA fragmentation compared with paracetamol challenge alone (p < 0.01) and this appeared also on gel electrophoresis analysis.
; B-tx «acute toxic» model plus DTS 150 mg/kg; C: «sub-toxic» model (150 mg/kg); C-tx: «sub-toxic» model plus DTS 150 mg/kg. Left panel: electrophoresis picture of DNA fragmentation following paracetamol intoxication. The fragmentation was markedly reduced in DTS-treated animals as calculated in the right panel. *p < 0.01 vs control values **p < 0.05 vs baseline values")
DNA fragmentation after paracetamol intoxication and DTS treatment.
Group A: control: B: «acute toxic» model (1.5 g/kg); B-tx «acute toxic» model plus DTS 150 mg/kg; C: «sub-toxic» model (150 mg/kg); C-tx: «sub-toxic» model plus DTS 150 mg/kg.
Left panel: electrophoresis picture of DNA fragmentation following paracetamol intoxication. The fragmentation was markedly reduced in DTS-treated animals as calculated in the right panel.
*p < 0.01 vs control values
**p < 0.05 vs baseline values
Paracetamol is widely used as an analgesic and antipyretic agent. However, accidental or intentional intake of high doses often causes acute hepatocellular necrosis with high morbidity and mortality.26,27 It is generally acknowledged that the toxicity of paracetamol is mediated by cy-tochrome P450 to generate a rather toxic metabolite, N-acetyl-p-benzoquinone imine, whose detoxification may lead to a dramatic depletion of hepatic GSH as also appeared from our study at sub-toxic dosage. Furthermore, NAPQI covalently binds to cysteine residues on proteins, resulting in the formation of 3-(cysteine-S-yl) paracetamol adducts. Consequently, N-Acetyl-cysteine, as a precursor of GSH, represents the most accepted standard therapeutic approach for paracetamol-induced acute liver injury.28 Studies with mouse hepatocytes culture have demonstrated that the oxidant stress, as measured by increased 2’, 7’-dichlorodihydrofluorescein diacetate fluorescence, substantially precedes cell injury by several hours,7 due to an early increase in the GSSG/GSH ratio.6 Although, a time-course monitoring was beyond our study, it was interesting to note that, an impaired redox balance in liver tissue was a remarkable feature, irrespective of the extent of paracetamol dosage. The administration of DTS proved to significantly limit GSH depletion, although the detailed mechanisms await further studies. During the previously mentioned redox imbalance, mitochondria are the primary targets in acetaminophen toxicity29 and oxidants such as peroxides and peroxynitrite, Ca2+, and Pi determines a mi-tochondrial permeability transition (MPT). This triggers an abrupt increase in the permeability of its inner membrane, the uncoupling of oxidative phosphorylation and the release of intramitochondrial ions and metabolic intermediates with mitochondrial damage.30 This phenomenon represents a central event in apoptosis and MPT is a lethal event for the cell biology by generating itself increased oxidant stress. In our present study, it was observed that administration of DTS caused a significant increase in activities of enzymatic and non-enzymatic antioxidant enzymes in the paracetamol damaged liver of rats, which sig-nificantly reduced the tissue level of MDA, thus lessening the cytolitic hepatocyte markers. Moreover, the generation of ROS by either paracetamol metabolism or resulting mitochondrial damage can lead to direct or indirect oxida-tive DNA damage and it has been found that a significant accumulation of the potentially mutagenic DNA lesion either at sub-toxic doses of the drug.31 We didn’t address the issue of measuring free radicals-damaged DNA products but it came clear that DNA fragmentation was a very prominent feature not only at toxic dosage of paracetamol in agreement with Ray et al.32 but also at sub-toxic poisoning while DTS proved to significantly reduce such phenomenon. Knight et al.6 has provided convincing evidence that, despite mitochondrial oxidant stress and peroxynitrite formation after paracetamol overdose, lipoperoxidation is not the major pathophysiological event. This would explain why, lipid-soluble antioxidants are ineffective in reducing this specific liver injury.33 On the other hand, water-soluble radical scavengers and/or redox epigenetic modula-tors, as it would seem DTS to be regarded, may be more promising as therapeutic agents in preventing the progression of paracetamol-induced hepatic injury. Indeed, the present study has demonstrated that DTS supplementation actively prevents the severe impairment of membrane-bound ATPase activity and protein thiols which are indeed physiological free radical scavengers. Indeed, profound changes in cell energy metabolism with a marked loss of total ATPase has been observed in animals administered paracetamol alone at whatever dosage in our study, which might be due to the loss of protein SH groups. This impairment is due to the alkylation of membrane proteins by reactive paracetamol metabolites. In particular, calcium AT-Pase activity within the plasma membrane which is an important regulating factor to maintain low cytosolic calcium levels and its activity is inhibited by the loss in free sulphydryl groups, whether by alkylation or oxidation. This is because the drug-induced limitation of the mitochondria to buffer a non-physiological increase in calcium may lead to the inappropriate stimulation of a number of calcium-activated catabolic enzymes inappropriate stimulation of a number of calcium-activated cata-bolic enzymes. Thus, intracellular calcium homeostasis is of great importance to cell viability and a variety of toxicant-induced hepatocellular injury results in the influx of Ca2+ into the cell, giving rise to a cascade of toxic events and resulting in cell death. It is suggested that, at low concentrations, NAPQI induces calcium release mainly via its oxidizing properties, which result in pyridine nucleotide hydrolysis and the stimulation of protein mono ADP ribo-sylation while at high concentrations protein arylation may also be a contributing factor. Although we didn’t examine the whole set of inorganic cations in liver tissue, it appeared that also animal intoxicated with even sub-toxic dosages of paracetamol, show elevated levels of calcium in the liver tissue (• •60%) and this abnormality was prevented when administered with DTS. This overall protective effect may be due to the presence of some antiapop-totic-and (water-soluble) antioxidant-endowed compo-nents34-37 which may have prevented the excessive oligonucleosomal DNA integrity in the face of paracetamol challenge. Besidethe direct quenching activity against metabolic activation of reactive metabolites, a protective epigenomic effect modulating the generesponsible for synthesiof antioxidant enzymeand DNA repair ilikely to have taken place and it the matter of currently ongoing research. Moreover, a limitation in our present study warranting further research irepresented by the need to ascertain whether DTS directly inhibited the metabolism of paracetamol to the toxic metabolite NAPQI while caution hato be applied in consideration of possible specie-specific differencetoo.
Nonetheless, taken overall, these data show that even sub-toxic dosage of paracetamol can initiate significant injurious hepatocyte damage and that DTS supplementation might be a promising clinically-applicable integra-tive approach whenever drug therapy is required for prolonged time and, most important, in those subjects who are already under poly-drug treatment.



