




Hepatocellular carcinoma is a lethal disease that requires a multidisciplinary approach and management. Surgical therapy offers long-term survival; however, few patients are candidates. There has been no accepted systemic therapy for this disease until recently. This article briefly discusses the role of RAS/RAF/MEK/ ERK signaling pathway in the pathogenesis of the disease and the promising role of sorafenib for advanced disease.
List abbreviations:
HCC, hepatocellular carcinoma
ERK, extracellular signal-regulated kinase
MEK, extracellular signal-regulated kinase
HBV, hepatitis B virus
HCV, hepatitis C virus
TKRs, tyrosine kinase receptors
GEFs, guanine nucleotide exchange factors; SOS protein, mammalian homologue of the Drosophila son of sevenless gene product
VEGF, vascular endothelial growth factor
PDGF, platelet-derived growth factor
EGF, epidermal growth factor
VEGFRs, VEGF receptors; HSP, heat shock protein
Hepatocellular carcinoma (HCC) is the most common primary liver cancer, with an estimated incidence of half a million new cases per year around the world.1 Its asymptomatic development, malignant progression, and the poor efficacy of current treatments entail a poor prognosis, with fewer than 5% of patients surviving five years after diagnosis. HCC is the third greatest cause of cancer- related death in the world, and most of these deaths are registered in developing countries.2
Intense research over the past 20 years has provided detailed information about the molecular mechanisms and signaling pathways involved in hepatocarcinogenesis. The RAS/RAF/MEK/ERK signaling pathway has an essential role in the regulation of normal hepatocyte proliferation. Defects in this signaling pathway are critical in HCC pathogenesis,3 making it an attractive target for chemotherapeutic agents. Sorafenib, an oral drug developed as a RAF inhibitor, is a promising agent for HCC therapy. Sorafenib is a multikinase inhibitor targeting the RAF/MEK/ERK pathway, with antiangiogenic effects.4,5 This review summarizes current knowledge of the RAS/RAF/MEK/ ERK signaling pathway and its implications in HCC pathogenesis, and focuses on the role of sorafenib in the therapy of HCC.
Epidemiology and risk factorsLiver-cancer-related death is a major health problem around the world. Despite being the sixth most common malignancy, HCC is highly lethal, representing the third greatest cause of death from malignancy worldwide, particularly in association with hepatitis B virus (HBV) infection in developing countries.3
The age-adjusted incidence of HCC has marked geographic variations, with the highest rates being observed in Asia and sub-Saharan Africa (25 cases per 100,000), and the lowest in North America and northern Europe.4,7 Unfortunately, several studies have shown a worldwide rise in HCC incidence in the last two decades, forecasting a devastating effect if health care policies are not intensified. Capocaccia et al.8 analyzed the database of the Surveillance Epidemiology and End Results program of the EUROCARE project and observed a fourfold increase in the incidence of HCC in southern Europe. In the United States, several studies have reported that the age-adjusted incidence of HCC has doubled over the last two decades,6-8 affecting Caucasian and Hispanic men particularly.9 At least 50% of the new cases in United States could be attributable to chronic Hepatitis C Virus (HCV) infection.10 However, in almost 15%- 50% of patients with HCC, there is no evidence of either viral hepatitis or heavy alcohol consumption,11 suggesting that such cases could be linked to nonalcoholic fatty liver disease and other etiologies of chronic liver disease.
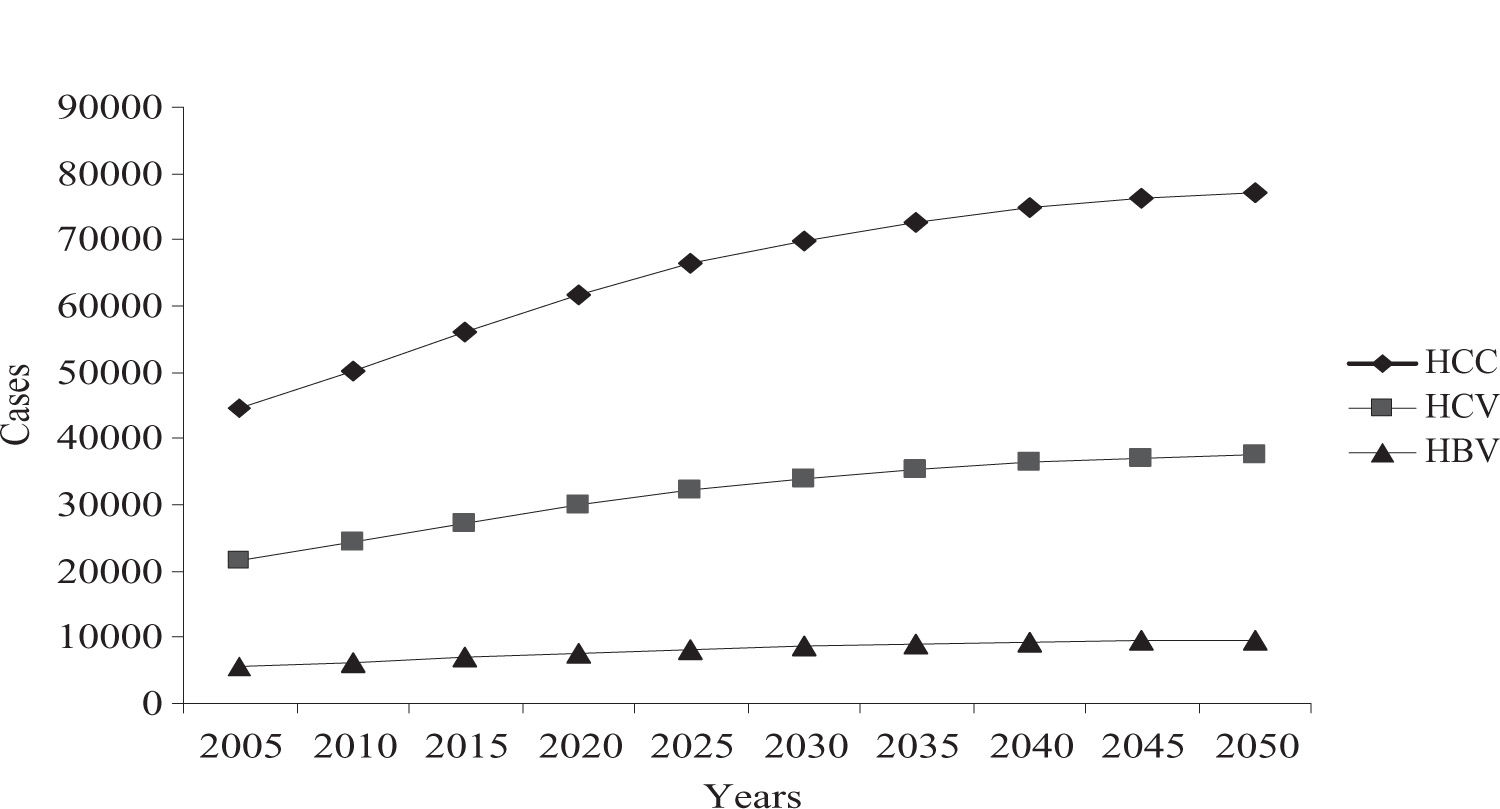
A preexistent cirrhotic liver is a clinicopathological condition observed in 80%–90% of patients who develop HCC.12 Several studies have indicated that 1%–4% of all cirrhotic patients per year will developed HCC,13 with differences according to the leading cause (table I).14-18 Liver cirrhosis has a critical impact on public health in Mexico, representing the third greatest cause of death in the general population, and predicted trends for the next five decades are not promising (figure 1).19,20 The Mexican Association of Hepatology determined alcohol and HCV infection to be the main causes of liver cirrhosis in Mexico (39.5% vs 36.6%, respectively; p = 0.113), followed by cryptogenic cirrhosis.21
Annual incidence of hepatocellular carcinoma related to the etiology of liver cirrhosis.
| Author, year | No. Patients | Etiology of liver cirrhosis | Mean Follow-up* | HCC Annual Incidence** |
|---|---|---|---|---|
| Fattovich et al., 1997 | 361 | HCV | 60 | 1.4% |
| Chiaramonte et al., 1999 | 166 | HCV | 64.5 | 3.8% |
| Fattovich et al., 2002 | 136 | HCV | 66 | 2.5% |
| Solá et al., 2006 | 200 | HCV | 39 | 5.5% |
| 177 | Alcoholic | 39 | 1.7% | |
| Fattovich et al., 2002 | 161 | HBV | 66 | 2.2% |
| Chiaramonte et al., 1999 | 66 | HBV | 64.5 | 1.7% |
| 27 | HBV/HCV | 64.5 | 7.6% | |
| Ratziu et al., 2002 | 22 | Cryptogenic, obesity-related | 18 | 0.8% |

Hepatocarcinogenesis is a multistep process in which genetic abnormalities and epigenetic alterations accumulate, causing aberrant growth and malignant transformation of hepatocytes. Accumulation of such abnormalities leads to activation of mediators of cellular proliferation (proto-oncogenes and their mitogenic signaling pathways) resulting in neoplastic potential.
Hepatocellular carcinomas exhibit a high degree of genetic heterogeneity, and multiple molecular pathways may be involved in the pathogenesis of HCC. The best- characterized pathways are the heat shock protein (HSP)/ stress response signaling pathway, the Wnt pathway, and the MAPK pathway, and the associated involvement of growth factors and cytokines.22-24 This review focuses on the MAPK pathway (the RAF/RAS/MEK/ERK signaling pathway) and its major role in hepatocarcinogenesis.
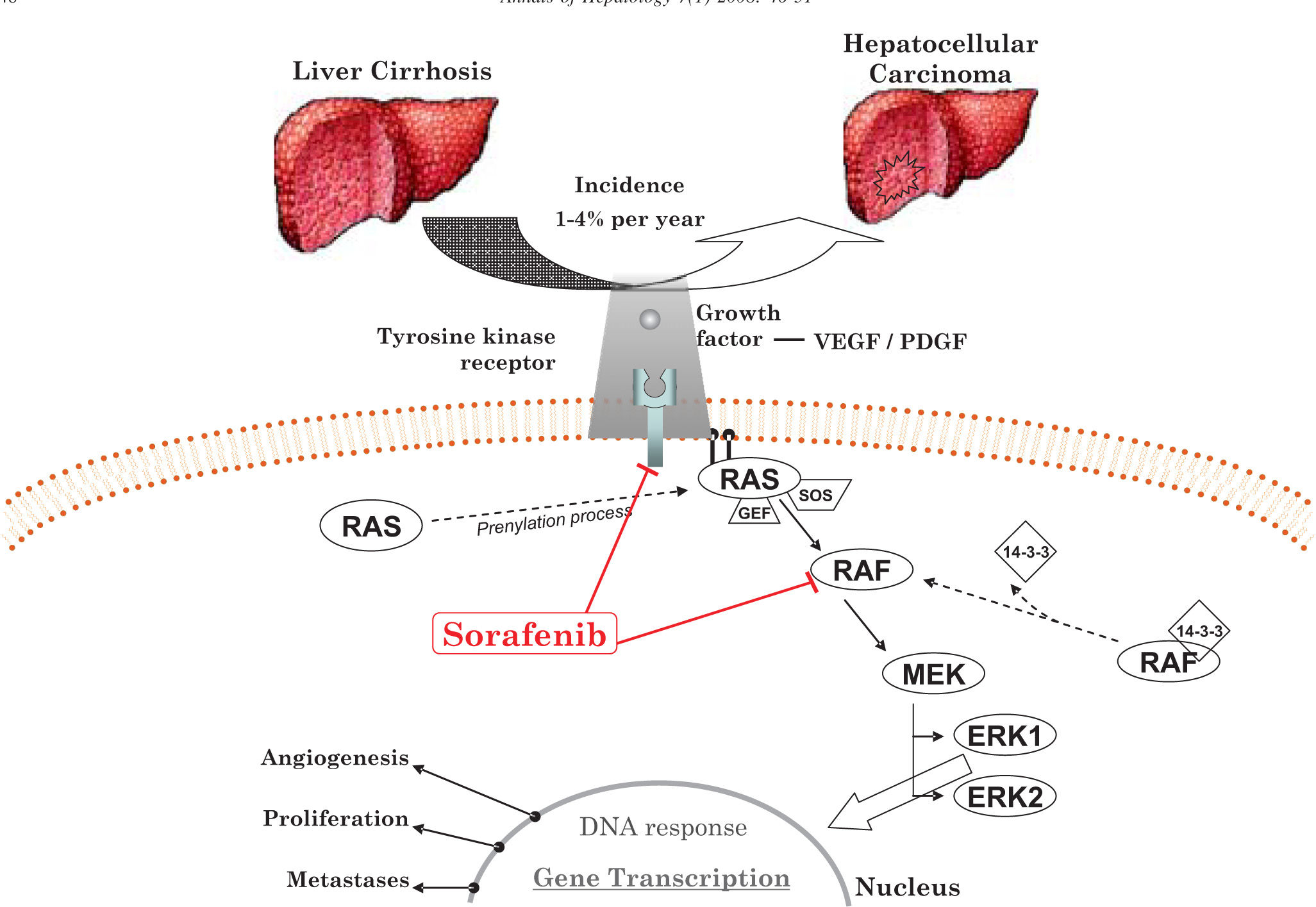
The RAS/RAF/MAPK-ERK signaling pathwayThe RAS/RAF/MAPK-ERK signaling pathway is an important mediator of tumor cell proliferation, differentiation and apoptosis. Studies have reported that MAPK expression is significantly higher in HCC compared to adjacent normal liver cells,25,26 showing the critical role of this pathway in the pathogenesis of HCC (figure 2).

RAS is a cytosolic protein that, after a prenylation process, is located on the inner surface of the cellular membrane. This posttranslational processing anchors RAS protein to the cytoplasmic membrane, which is necessary for its biological activity. Misplaced RAS proteins are inactive, probably because they cannot recruit their target enzymes. Interestingly, a recent study suggested that prenylation is not necessary for endogenous RAS activation in normal cells.27,28
Activation of RASActivation of the normal RAS signaling pathway is initiated by the interaction of several cytokines, hormones and extracellular growth factors with their tyrosine-kinases receptors (TKRs). As a result, ligand binding induces receptor dimerization and autophosphorylation, activating downstream intracellular signal cascades. First, there is recruitment of guanine nucleotide exchange factors (GEFs), such as RAS-GRF and SOS protein (mammalian homologue of the Drosophila son of sevenless gene product), to the inner surface of the cell membrane where RAS protein is also located after prenylation. RAS is a membrane-bound G protein. The biological activity of RAS is regulated through the GDP/GTP cycle.29 In the inactive state, RAS exists in the GDP-bound form. Because of TKR activation, GEF is recruited and located in the cell membrane, promoting the formation of the GTP-bound active state. In contrast, RAS becomes inactivated through hydrolysis of GTP by an intrinsic GTPase. Nevertheless, in vitro studies have demonstrated a low-activity level of this intrinsic GTPase; thus, effective hydrolysis of GTP is performed by several cytoplasmic GTPase-activating proteins, which rapidly induce the hydrolysis of GTP-bound RAS to the inactive GDP form.
Unregulated RAS pathway activity is observed in tumor cells, because of point mutations in the RAS gene family and the overexpression of TKRs and their ligands. The RAS genes encode four highly similar 21 kDa proteins, H-RAS, N-RAS, K-RAS4A and K-RAS4B. Point mutations in RAS genes are observed in approximately 20%-30% of all solid tumors,29 K-RAS being the most commonly affected. For example, K-RAS is mutated in up to 80% of pancreatic adenocarcinomas.30 N-RAS mutations are observed in 30% of cases of HCC;31 in contrast, H-RAS mutations are rarely observed. These genetic derangements compromise the intrinsic GTPase activity and the GAP-induced GTPase activation of RAS,32 with the loss of its ability to return to a quiescent state, leading to constitutive activation of RAS and subsequent stimulation of downstream effectors.
Overactivation of RTKs results from activating mutations or overexpression of growth factor ligands. Vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) have important roles in dysregulated cell growth and metastases.33 VEGF and its receptors (VEGFRs), particularly VEGFR-2, are key molecules involved in endothelial cell proliferation, angiogenesis and vascular permeability.34 Elevated serum VEGF levels are associated with poor prognosis in patients with HCC.35,36 Furthermore, VEGF gene polymorphisms have been suggested as prognostic indicators for HCC.37 Experimental studies in mice have reported that approximately 70% of HCCs show high serum PDGFR-a levels.38
Receptor overexpression and elevated ligand availability play an important role in the development of metastases in patients with HCC. As VEGFR and PDG- FR use the RAS/RAF/MEK/ERK pathway, its targeting as an anticancer therapy has been explored with great interest.
RAF is the best downstream effector of RASRAF, the best-characterized downstream effector of RAS, is a serine/threonine protein kinase positioned as the first signaling element of the MAPK pathway.39,40 The RAF gene family encodes three closely related cytosolic proteins: ARAF, BRAF and CRAF (also termed RAF-1). Whereas each isoform has a distinct expression profile in tissues, the three RAF species are found in normal liver cells and recent evidence has shown that they are overexpressed or mutated in HCC.41,42 These serine/threonine protein kinases share three conserved sequence regions, termed CR1, CR2 and CR3, that participates in the complex processes of RAF kinase activity regulation.
To activate RAF kinase, GTP-bound RAS directly interacts with RAF, promoting its recruitment to the cell membrane, an essential step for its activation.43,44 The RAS effector domain binds to RAF via CR1. Once located in the membrane, RAF requires several modifications to become active.45 Nevertheless, in vitro studies have revealed that such an interaction is insufficient to stimulate RAF kinase activity, suggesting that others cofactors are necessary for RAF activation in vivo.46,47
Cytosolic CRAF exists as a complex formed with the dimeric cofactor 14-3-3, a highly conserved chaperonin protein that binds at phosphorylated serine residues S259 and S621, inactivating the protein. CRAF interaction with RAS displaces the cofactor 14-3-3, exposing the serine residues to desphosphorylation by protein phosphatase 2A or other phosphatases. In addition, the activation of CRAF also requires phosphorylation of other serine and tyrosine residues, particularly S338 and Y341.48 In BRAF, the final phosphorylation of S338 and Y341 is omitted because S445, a homologous site on BRAF, is constitutively phosphorylated; thus, BRAF is immediately activated after interaction with GTP-bound RAS. This property confers to BRAF a higher kinase activity level, making it the strongest activator of downstream MEK pathway.
Once activated, RAF phosphorylates MAPKKs (MEK1 and MEK2) at residues S218 and S222. All RAF isoforms are able to activate MEK1; however, only BRAF and CRAF activate MEK2.42 In turn, downstream effectors of MAPKKs, ERK1 and ERK2, are activated by phosphorylation at residues T183 and Y185, with further activation of the nuclear transcription factors Elk-1, fos, jun, AP-1, myc and nuclear factor-kB (NF-kB), which regulate gene expression associated with cell proliferation, differentiation, angiogenesis or apoptosis.49
Constitutive activation of RAF and RAS are indistinguishable in their potential to induce malignant transformation. RAF protein is mutated in approximately 7% of all malignancies because of point mutations, deletions, amplification and rearrangements of RAF. Tannapfel et al. recently reported that BRAF mutations are rare in HCC.50 Hwang et al. have shown that the CRAF gene is upregulated in 40% of cirrhotic livers and 50% of HCCs; as a consequence, CRAF protein is overexpressed in 91.2% and 100% of these tumors, respectively.51
Epidemiological data have provided strong evidence about the role of chronic HCV infection and cirrhosis in the development of HCC.52 In addition, experimental models have shown the development of HCC in transgenic mice expressing the HCV core gene.53 However, the precise mechanisms involved are unclear. An in vitro study showed that HCV core protein activates the MEK/ERKs signaling pathway in mammalian epithelial cells, with constitutive RAF-1 activity. HCV core protein has also been shown to bind the 14-3-3 protein both in vivo and in vitro, suggesting that such an interaction with this chaperonin exposes the serine residues S259 and S621 to desphosphorylation, a key step in RAF activation.54
Sorafenib: a promising therapySorafenib, a bi-aryl urea, was initially recognized as a CRAF inhibitor.4 Further studies in different cell lines and xenograft models have demonstrated that sorafenib is a potent multikinase inhibitor, including wild-type and mutant BRAF, VEGFR2, VEGFR3, PDGFR-***entity***, FLT3, Ret and c-Kit, and has antiangiogenic effects.55-57
The direct effects of sorafenib have been evaluated in vitro in two distinct HCC cell lines. Sorafenib inhibited cell proliferation and induced cell apoptosis in both cell lines in a dose-dependent manner.57 In addition, sor- afenib inhibited MEK and ERK phosphorylation. In the same study, the in vivo effects of sorafenib were evaluated in a xenograft model, in which sorafenib produced significant and dose-dependent tumor growth inhibition of 49% and 78%, respectively. Sorafenib produced durable partial tumor regression in 50% of the mice, indicating direct effects on tumor cell proliferation/survival in vivo.57
Two open-label, uncontrolled, phase I trials evaluated sorafenib in 86 patients with solid tumors refractory to standard treatment, including one patient with HCC.58,59 Overall, sorafenib was safe and well tolerated at doses of 400 mg b.i.d. Even when the majority of patients experienced at least one adverse event, toxicities were mostly mild to moderate. Two phase I clinical trials demonstrated efficacy in the treatment of patients with advanced HCC using combination regimens with other anticancer agents such as doxorubicin.60,61 Patients received continuous oral sorafenib 400 mg b.i.d. in four-week cycles (median number of treatment cycles was four, range 1-19). Three patients had partial responses (duration ranged from 12 to 14.5 months), eight had minor responses, 46 had stable disease (***entity***16 weeks) and 48 had progressive disease (imaging assessment), with a median overall survival of 9.2 months. In addition, relatively infrequent dose-limiting toxicities were observed in this study, including fatigue (9.5%), diarrhea (8%) and hand and foot skin reactions (5.1%), with grade 3 toxicities the most common.61
Results from the international double-blind placebocontrolled SHARP trial were presented during the Annual Meeting of the American Society of Clinical Oncology.62 After stratification for portal vein and/or extrahepatic invasion and ECOG status, the researchers randomized 602 patients with Child–Pugh A cirrhosis and HCC to receive either placebo (n = 303) or sorafenib 400 mg b.i.d. (n = 299) from March 2005 to April 2006. Intention-to- treat analysis revealed that sorafenib-treated patients lived 46.3 weeks as compared to 34.4 weeks in the placebo group (p = 0.00058). In addition, time-to-progression was significantly longer in the sorafenib-treated group (24 vs 12.3 weeks; p = .000007). No complete response was observed during the study period and few partial responses were observed (7/299 in the sorafenib-treated group vs 2/303 in the placebo-treated group). The study was stopped in February 2007 because of results favoring sorafenib were found in the second planned interim analysis of October 2006.62 Based on these final data, sorafenib seems to have a role as a disease stabilizer rather than as a cure for HCC.
ConclusionsMolecule-targeted therapies for cancer are promising, particularly those directed to malignancies with a currently poor prognosis, such as HCC. The available literature shows for the first time that systemic therapy with sorafenib prolongs survival in HCC patients. We anticipate great interest in the publication of the full report of the SHARP trial and its impact on the scientific community worldwide.



