




Liver fibrosis is a common pathological change in many chronic liver diseases. Activation of hepatic stellate cells (HSCs) is the core event in liver fibrosis. This study aimed to investigate the role of testicular orphan receptor 4 (TR4) in the activation of HSCs.
Materials and MethodsIn vivo, bile duct ligation (BDL)-induced rat liver fibrosis model was established, and the expressions of TR4 and α-smooth muscle actin (α-SMA) in liver tissues were detected. In vitro, TR4 knockdown and overexpression in JS-1 cells using lentiviral vectors were constructed, and the expressions of TR4, α-SMA, Col-I, and TGF-β1/smads and retinoid X receptor (RXR) pathway-related genes were detected.
ResultsTR4 was highly expressed in BDL-induced fibrotic liver, accompanied by increased expression of α-SMA. Knockdown of TR4 significantly inhibited the expressions of α-SMA, Col-I, p-TβRI, and p-Smad2/3, and up-regulated the expression of RXRα in HSCs in vitro. In contrast, TR4 overexpression significantly increased the expressions of α-SMA, Col-I, p-TβRI, and p-Smad2/3, and inhibited the expression of RXRα.
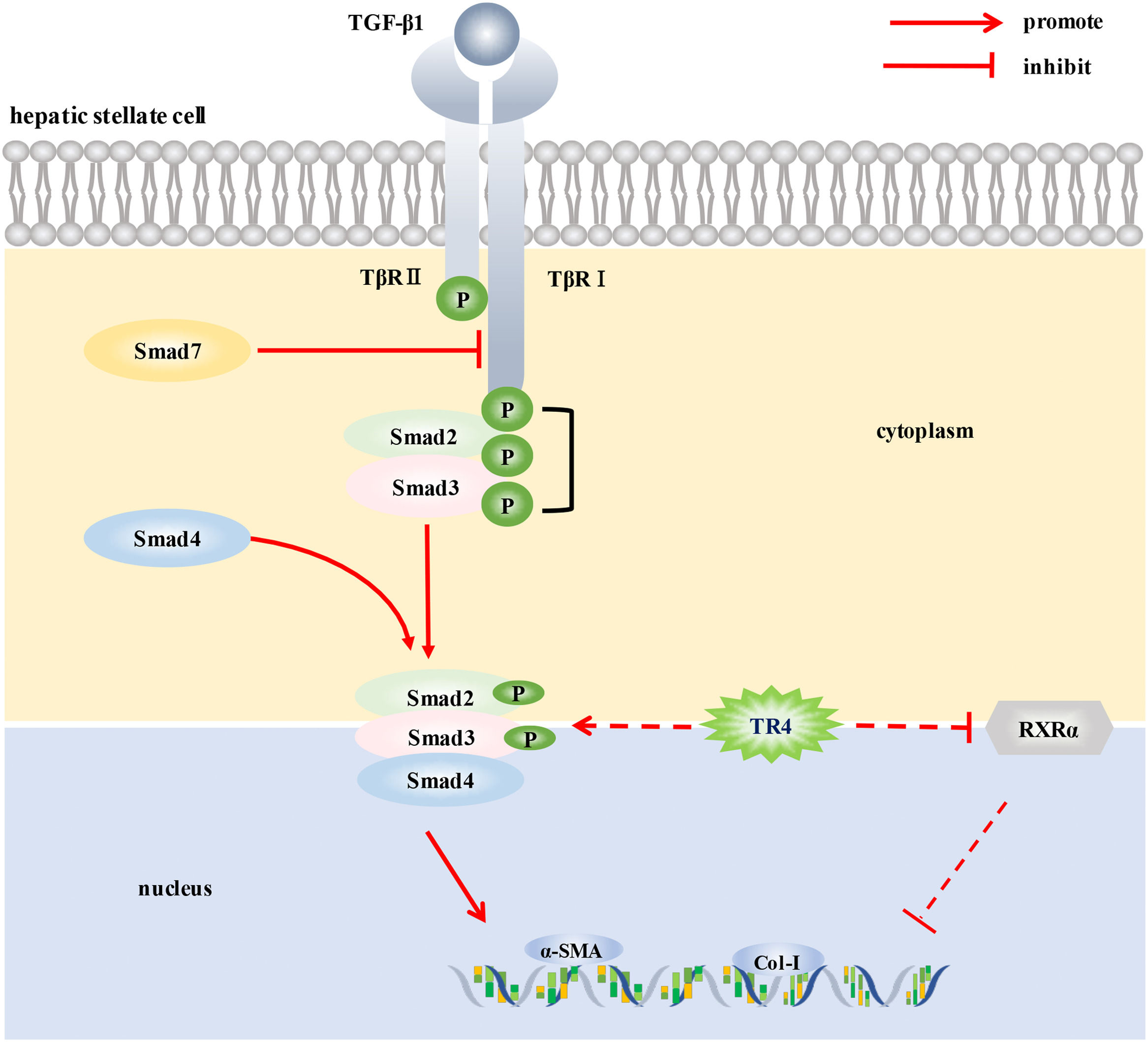
ConclusionsTR4 may promote the activation of HSCs by up-regulating TβR I/Smad2/3 signaling pathway and down-regulating RXRα signaling, thereby promoting the progression of liver fibrosis. Our findings may provide a new therapeutic target against hepatic fibrosis.
Liver fibrosis is a consequence of chronic liver injury caused by a wide range of pathogenic stimuli. Liver fibrosis is associated with a high risk of cirrhosis, liver cancer, and liver failure, for which the only existing potential treatment is liver transplantion [1]. Therefore, developing novel therapeutic strategies for preventing and treating liver fibrosis is a key imperative.
Activation of hepatic stellate cells (HSCs) in response to liver injury is a prerequisite for initiating liver fibrosis. Activation of HSCs is characterized by a switch from a quiescent vitamin A-rich phenotype to a proliferating, matrix-producing myofibroblastic fibrogenic phenotype [2]. Activated HSCs are the main source of myofibroblasts, which exhibit characteristics of proliferation, contraction, migration, secretion of extracellular matrix protein, and pro-inflammatory responses. Transforming growth factor β1 (TGF-β1)/smads signaling pathway is considered the most important signaling pathway for the activation of HSCs [2]. Studies have shown that a number of injury-related factors may promote the transdifferentiation of HSCs and the proliferation of myofibroblasts. Conversely, the resolution of injury led to gradual inactivation and involution of myofibroblast populations and a tendency for regression of liver fibrosis [3].
Testicular orphan receptor 4 (TR4) belongs to the nuclear receptor superfamily, encoding a 67-kDa protein. TR4 was initially isolated from the cDNA library of the hypothalamus, prostate, and testis in humans and rats; structurally, it is a member of the orphan nuclear receptor family [4]. Due to the specific ligands, the agonists and inhibitors of TR4 have not yet been discovered and there has been slow progress in the early study of TR4. The successful construction of TR4 knockout (TR4−/−) mice in recent years has helped unravel many important physiological functions involving TR4, such as fertility, neuronal development, lipid metabolism, and bone development [5]. TR4 was shown to promote the transformation of prostate epithelial cells, and enhance the invasion and metastasis of prostate cancer cells, a phenomenon that was related to the regulation of TGF-β1/smads signaling pathway [6,7]. As mentioned above, TGF-β1/smads is also the most important signaling pathway implicated in promoting the activation of HSCs. We speculate that TR4 may also potentially affect the activation of HSCs by regulating the TGF-β1/smads signaling pathway.
2Materials and Methods2.1Animals and experimental protocolAdult Sprague–Dawley (SD) male rats (160∼180 g, specific pathogen-free grade) were purchased from Shanghai Xipuer-Bikai Experimental Animal Co. Ltd. (Shanghai, China) and housed at the Laboratory Animal Center at the School of Pharmacy, Fudan University (Shanghai, China) in a controlled environment (ambient temperature 25±2°C, relative humidity 40-60%; 12/12 h light-dark cycle).
SD rats were randomly divided into the sham-operated group (Sham) (n=8) and the bile duct ligation group (BDL) (n=8). BDL surgery was performed as previously described [8]. At the end of the 4th week after the operation, all rats were anesthetized with an intraperitoneal injection of sodium pentobarbital; then, blood and liver samples were collected and stored at −80°C for subsequent testing. Liver tissues were harvested and fixed in 10% neutral-buffered formalin for pathological analysis.
2.2Sirius red staining of liver tissuesSirius red (SR) staining was performed using 4-μm thick paraffin-embedded liver tissue sections to evaluate the degree of liver fibrosis, as previously described [9].
2.3Quantitative Real-Time Polymerase Chain Reaction (qPCR)Total RNA in liver tissues or cells was isolated and reverse transcribed using Total RNA Purification Kit MagExtractor (Code: NPK-201, Toyobo Co. Ltd., Osaka, Japan) and the Rever Tra Ace qPCR RT Master Mix with gDNA Remover (Code: FSQ-301, Toyobo Co. Ltd., Osaka, Japan). GAPDH was used as the housekeeping gene for the normalization of the target gene expressions. The detailed procedure for qPCR is described elsewhere [8]. The primer sequences used are listed in Supplementary Table 1.
2.4Western blottingWestern blotting was performed as previously described [8]. 30∼50 μg proteins were used for immunoblot analysis using the Odyssey 2.1 software of the Odyssey infrared scanner (LI-COR Biosciences, Lincoln, NE, United States). The greyscale values were analyzed by ImageJ software, using GAPDH for normalization of the target protein expression. The antibodies used in this study are listed in Supplementary Table 2.
2.5Cell lines and lentiviral transfectionThe immortalized mice hepatic stellate cell line (JS-1) was obtained from the Institute of Liver Diseases, Shuguang Hospital, affiliated with the Shanghai University of Traditional Chinese Medicine. Lentivirus vector containing short hairpin RNA (shRNA) of TR4 gene knockdown (shTR4), shRNA of TR4 overexpression (OETR4) and empty control lentiviral (LV-control) was designed and produced by Genechem Co. Ltd., (Shanghai, China). JS-1 cells transfected with shTR4 or OETR4 lentiviral were obtained by transfection of the JS-1 cells with GV248 constructs that contained shTR4 cDNA and GV358 constructs containing OETR4 cDNA, respectively, followed by puromycin selection for 48 hours. The shRNA sequence was: CCAGACAATAGCAACTAAA, and the control sequence was: TTCTCCGAACGTGTCACGT. Cells were harvested for passage or testing after the attainment of 80% confluence.
2.6Statistical analysisAll data were expressed as mean ± standard deviation (SD) and analyzed using SPSS2.0 software. Differences between the two groups were assessed using a two-tailed Student's t-test. One-way analysis of variance followed by the least significant difference test was performed for multi-group comparisons. P values < 0.05 were considered indicative of statistical significance.
2.7Ethical StatementsAll animal experiments complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and were conducted in accordance with the UK Animals (Scientific Procedures) Act, 1986, and associated guidelines, EU Directive 2010/63/EU for Animal Experiments. All animal experiments were approved by the Experimental Animal Ethics Committee of the School of Pharmacy, Fudan University (No. 2018-07-SZYD-LP-01).
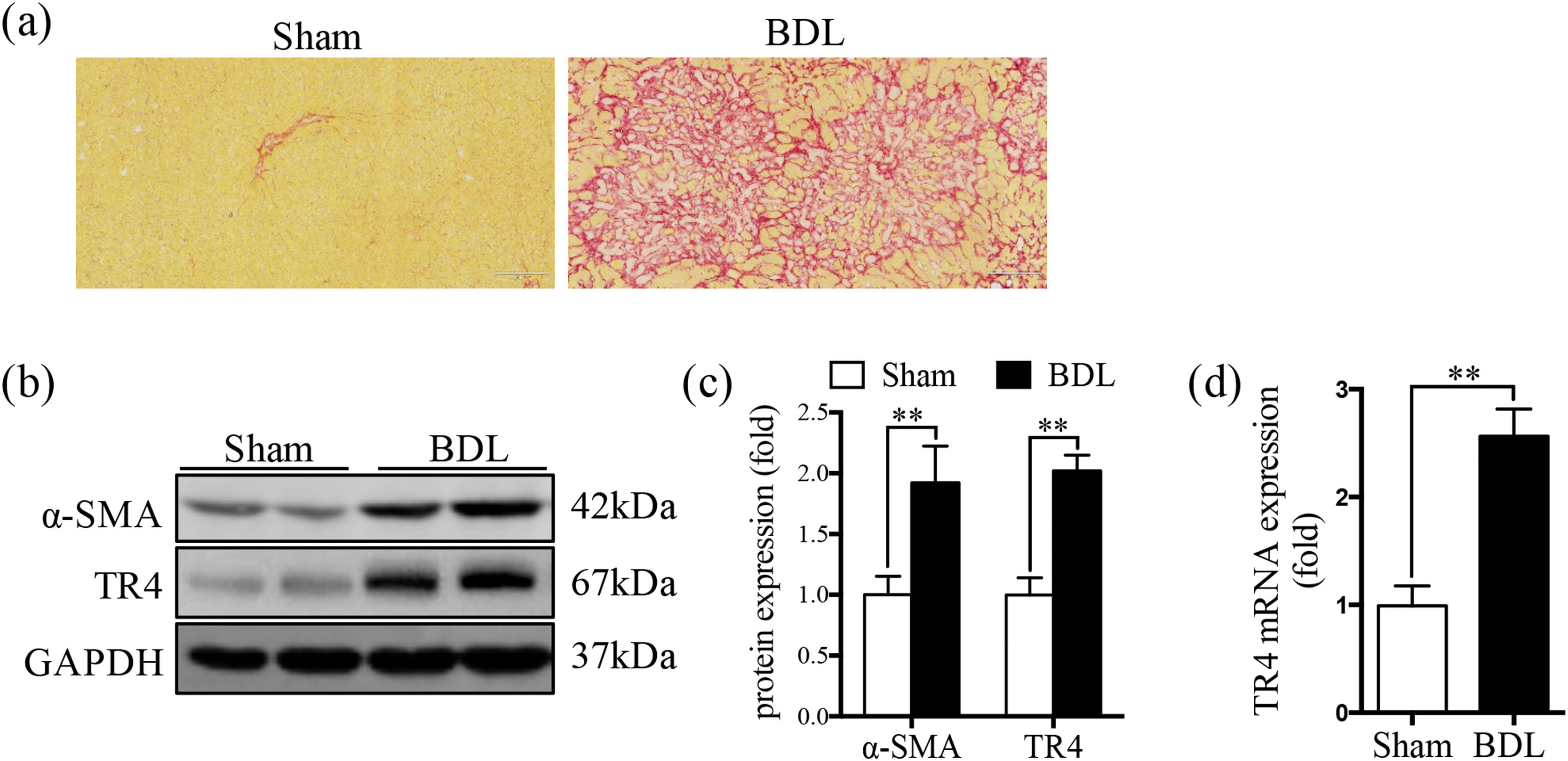
3Results3.1TR4 expression was significantly increased in BDL-induced fibrotic liverSR staining showed significantly greater collagen deposition in the liver tissue of the BDL rats compared to that in the Sham group (Fig. 1a). The protein expression of α-SMA was significantly elevated in the BDL group, accompanied by an increase in TR4 mRNA and protein expressions, compared to the Sham group (Fig. 1b-1d). These results indicated that the increased expression of TR4 in the context of liver fibrosis might be associated with the activation of HSCs.
 Representative images of liver sections stained by SR (100 ×). (b) Western blots showing the protein expressions of α-SMA and TR4. (c) Quantitative analysis of expressions of α-SMA and TR4, using GAPDH as loading control. (d) Results of qPCR showing gene expressions of TR4. Data are presented as mean ± SD (n = 8 per group). ⁎⁎P<0.01. Sham, the sham-operated group; BDL, the bile duct ligation group.")
Expression of TR4 was significantly increased in BDL-induced fibrotic liver. (a) Representative images of liver sections stained by SR (100 ×). (b) Western blots showing the protein expressions of α-SMA and TR4. (c) Quantitative analysis of expressions of α-SMA and TR4, using GAPDH as loading control. (d) Results of qPCR showing gene expressions of TR4. Data are presented as mean ± SD (n = 8 per group). ⁎⁎P<0.01. Sham, the sham-operated group; BDL, the bile duct ligation group.
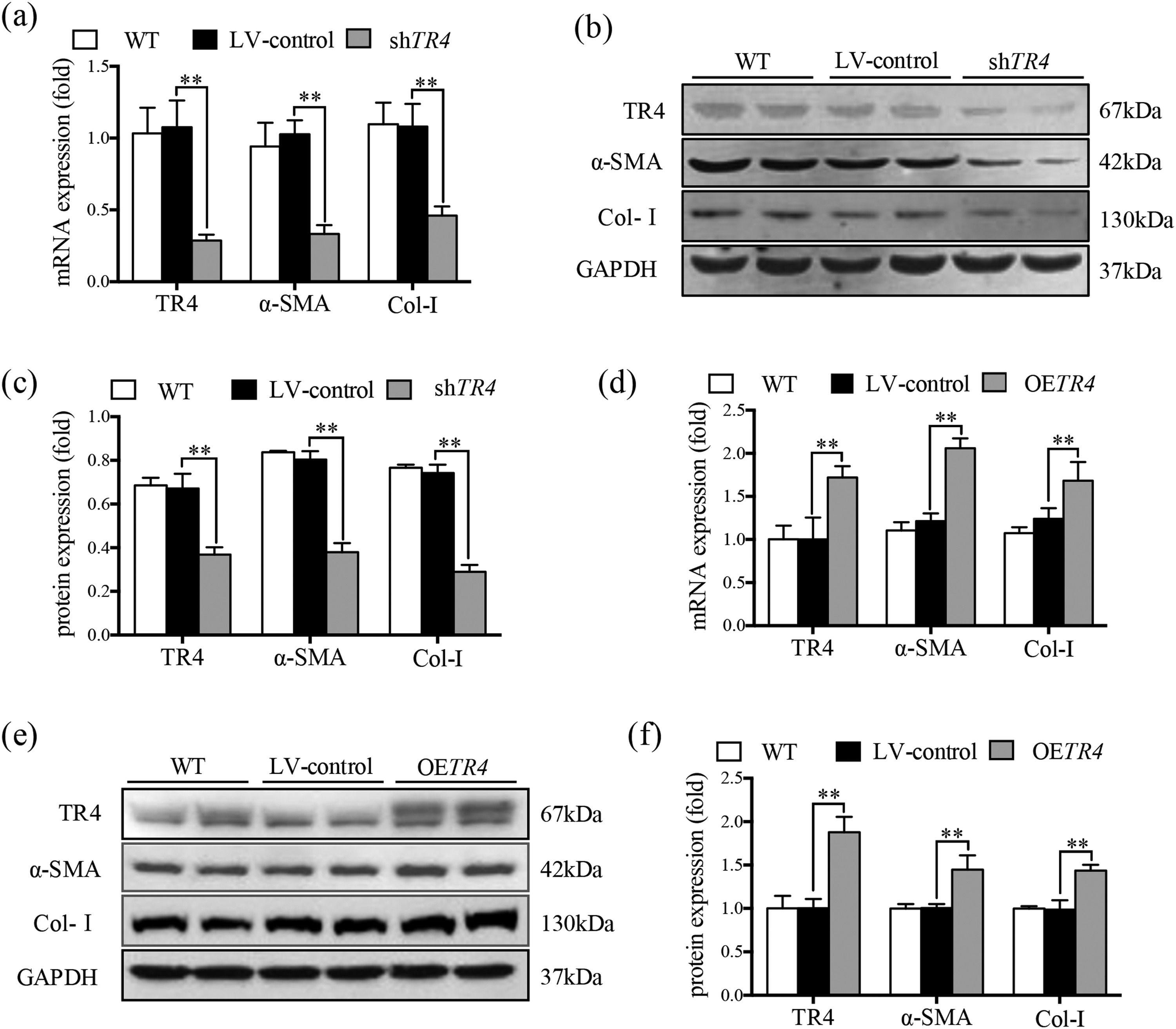
JS-1 cells were transduced with a lentiviral vector encoding shRNA against TR4 for its knockdown (ShTR4) to verify the role of TR4 in the activation of HSCs, qPCR and Western blot analysis revealed no significant differences between the without transfection (WT) group and LV-control group with respect to the expressions of TR4, α-SMA, or Col-I (Fig. 2a-2c). The expression of TR4 was effectively interfered in JS-1 cells, as the mRNA expression of TR4 was reduced by 79.2% in the shTR4 group, compared to the LV-control group. Moreover, the mRNA and protein expressions of α-SMA and Col-I in the shTR4 group were lower than those in the LV-control group (Fig. 2a-2c). In contrast, TR4-overexpressing cells (OETR4) showed remarkable upregulation of TR4, accompanied by increases in the expressions of α-SMA and Col-I (Fig. 2d-2f). These results indicated the important role of TR4 in the activation of HSCs.
 the mRNA expressions of TR4, α-SMA and Col-I determined by qPCR; (b) protein expressions of TR4, α-SMA and Col-I detected by Western blot (GAPDH was used as loading control); (c) quantitative analysis of expressions of TR4, α-SMA, and Col-I proteins. After overexpression of TR4 in JS-1 cells, (d) gene expressions of TR4, α-SMA and Col-I detected by qPCR; (e) protein expressions of TR4, α-SMA, and Col-I detected by Western blot; (f) quantitative analysis of TR4, α-SMA, and Col-I proteins. Data are presented as mean ± SD. ⁎⁎P<0.01. WT, the without transfection cells; LV-control, the control lentiviral cells; shTR4, the TR4 knockdown cells; OETR4, the TR4 overexpression cells.")
TR4 triggered the activation of HSCs. After TR4 knockdown in JS-1 cells, (a) the mRNA expressions of TR4, α-SMA and Col-I determined by qPCR; (b) protein expressions of TR4, α-SMA and Col-I detected by Western blot (GAPDH was used as loading control); (c) quantitative analysis of expressions of TR4, α-SMA, and Col-I proteins. After overexpression of TR4 in JS-1 cells, (d) gene expressions of TR4, α-SMA and Col-I detected by qPCR; (e) protein expressions of TR4, α-SMA, and Col-I detected by Western blot; (f) quantitative analysis of TR4, α-SMA, and Col-I proteins. Data are presented as mean ± SD. ⁎⁎P<0.01. WT, the without transfection cells; LV-control, the control lentiviral cells; shTR4, the TR4 knockdown cells; OETR4, the TR4 overexpression cells.
After transfection with shTR4 lentivirus, there was a significant decrease in the mRNA and protein expressions of TGF-β1; in addition, the mRNA expressions of TβRI, Smad 2 and Smad3 were reduced (Supplementary figure 1b). The ratio of p-TβRI to TβRI and ratio of p-Smad2/3 to Smad2/3 were significantly down-regulated, as detected by Western blot, while there was no significant change in the expression of Smad7 (Fig. 3a-3c). In contrast, the mRNA expressions of TβRI, Smad 2 and Smad3 (Supplementary figure 1a), and the ratio of p-TβRI to TβRI and the ratio of p-Smad2/3 to Smad2/3 were markedly increased in the OETR4 group compared to the LV-control group. Besides, there was no significant difference in these indices between the without-transfection group and the LV-control group (Fig. 3a-3e).
 protein expressions of p-TβRI, TβRI, p-Smad2/3, Smad2/3, TGF-β1, and Smad7 detected by Western blot; (b) quantitative analysis of p-TβRI, TβRI, p-Smad2/3, Smad2/3, TGF-β1, and Smad7 proteins;(c) mRNA expressions of TGF-β1 and Smad7 detected by qPCR. After overexpression of TR4 in JS-1 cells, (d) protein expressions of p-TβR1, TβR1, p-Smad2/3, Smad2/3, TGF-β1, and Smad7 detected by Western blot; (e) quantitative analysis of p-TβRI, TβRI, p-Smad2/3, Smad2/3, TGF-β1 and Smad7 proteins. Data are presented as mean ± SD. ⁎⁎P<0.01. WT, the without transfection cells; LV-control, the control lentiviral cells; shTR4, the TR4 knockdown cells; OETR4, the TR4 overexpression cells.")
TR4 regulated TGF-β1/Smads signaling pathway. After TR4 knockdown in JS-1 cells, (a) protein expressions of p-TβRI, TβRI, p-Smad2/3, Smad2/3, TGF-β1, and Smad7 detected by Western blot; (b) quantitative analysis of p-TβRI, TβRI, p-Smad2/3, Smad2/3, TGF-β1, and Smad7 proteins;(c) mRNA expressions of TGF-β1 and Smad7 detected by qPCR. After overexpression of TR4 in JS-1 cells, (d) protein expressions of p-TβR1, TβR1, p-Smad2/3, Smad2/3, TGF-β1, and Smad7 detected by Western blot; (e) quantitative analysis of p-TβRI, TβRI, p-Smad2/3, Smad2/3, TGF-β1 and Smad7 proteins. Data are presented as mean ± SD. ⁎⁎P<0.01. WT, the without transfection cells; LV-control, the control lentiviral cells; shTR4, the TR4 knockdown cells; OETR4, the TR4 overexpression cells.
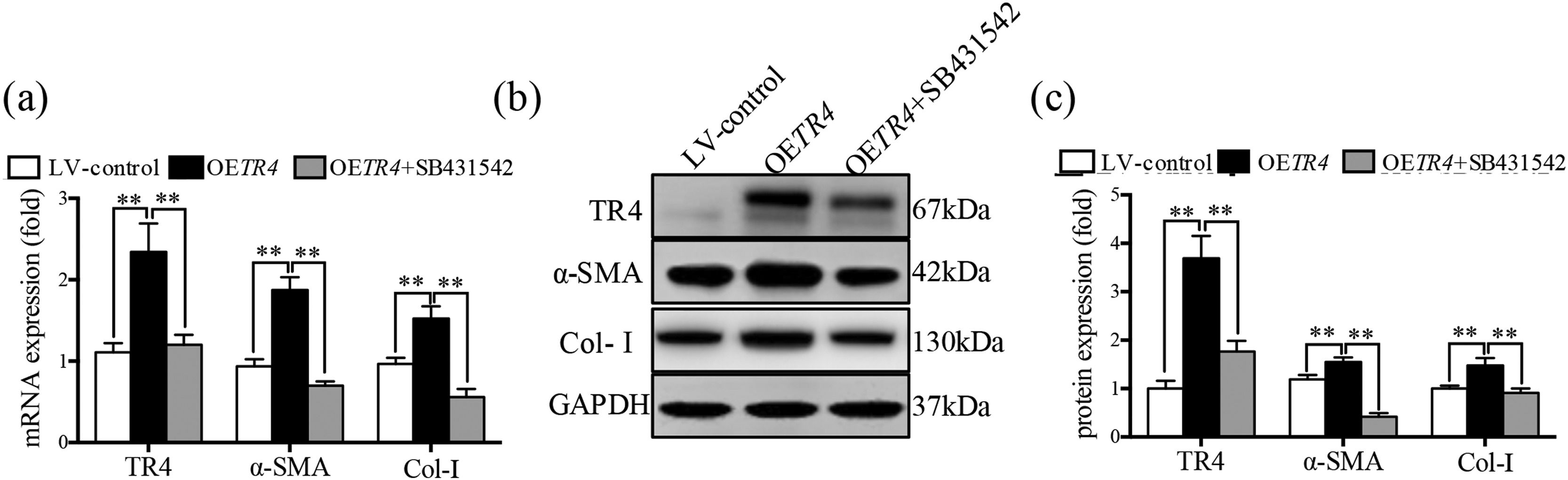
After transfection with OETR4 lentivirus in JS-1 cells, there was a significant increase in the expressions of α-SMA and Col-I. However, treatment of OETR4 transfected JS-1 cells with SB431542 (an inhibitor of TGF-β receptor) significantly decreased the expressions of TR4, α-SMA, and Col-I (Fig. 4a-4c). The results suggested that TR4-mediated activation of HSCs was suppressed by SB431542, which further confirmed activation of the TGF-β receptor after overexpression of TR4.
 mRNA expressions of α-SMA, Col-I, and TR4 detected by qPCR; (b) protein expressions of α-SMA, Col-I, and TR4 detected by Western blot; (c) quantitative analysis of α-SMA, Col-I, and TR4 proteins. Data are presented as mean ± SD. ⁎⁎P<0.01. LV-control, the control lentiviral cells; OETR4, the TR4 overexpression cells; OETR4+SB431542, the TR4 overexpression cells plus treatment with SB431542.")
The activation of HSCs induced by TR4 overexpression was counteracted after the inhibition of TGF-β receptor expression. After OETR4 JS-1 cells were treated with SB431542, (a) mRNA expressions of α-SMA, Col-I, and TR4 detected by qPCR; (b) protein expressions of α-SMA, Col-I, and TR4 detected by Western blot; (c) quantitative analysis of α-SMA, Col-I, and TR4 proteins. Data are presented as mean ± SD. ⁎⁎P<0.01. LV-control, the control lentiviral cells; OETR4, the TR4 overexpression cells; OETR4+SB431542, the TR4 overexpression cells plus treatment with SB431542.
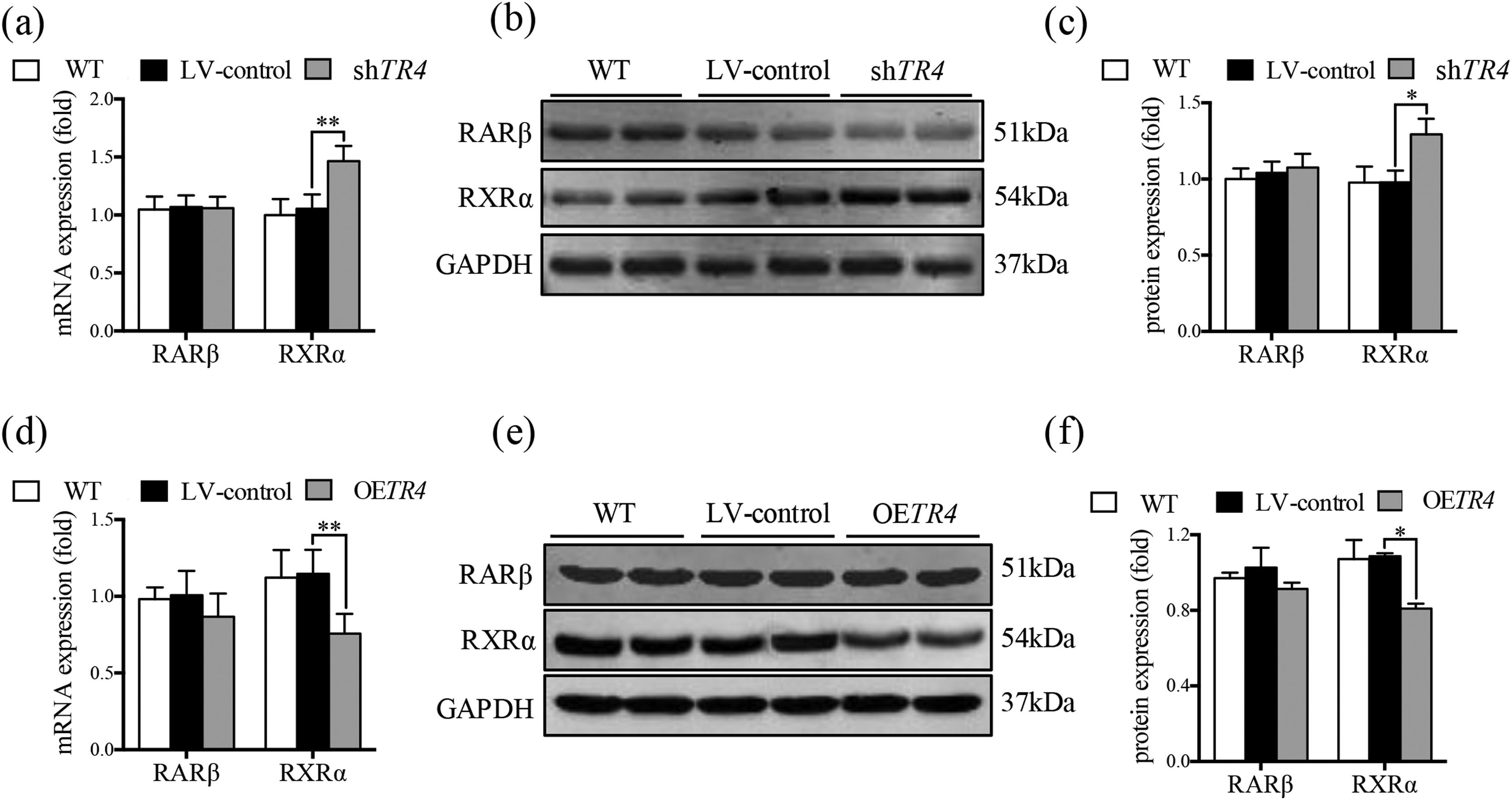
After TR4 knockdown in JS-1 cells, there was no significant difference between the LV-control group and without transfection group with respect to the protein and mRNA expressions of RXRα and retinoic acid receptor β (RARβ). The mRNA and protein expressions of RXRα in the shTR4 group were significantly increased as compared to those in the LV-control group. However, the expression of RARβ showed no significant change (Fig. 5a-5c). After overexpression of TR4 in JS-1 cells, the expression of RXRα was significantly down-regulated, while RARβ expression was still not changed (Fig. 5d-5f).
 mRNA expressions of RARβ and RXRα detected by qPCR; (b) protein expressions of RARβ and RXRα detected by Western blot; (c) quantification of RARβ and RXRα proteins. After lentivirus-mediated overexpression of TR4 in JS-1 cells, (d) mRNA expressions of RARβ and RXRα detected by qPCR; (e) protein expressions of RARβ and RXRα detected by Western blot; (f) quantification of RARβ and RXRα proteins. Data are presented as mean ± SD. *P<0.05, ⁎⁎P<0.01. WT, the without transfection cells; LV-control, the control lentiviral cells; shTR4, the TR4 knockdown cells; OETR4, the TR4 overexpression cells.")
TR4 regulated the expression of RXR α. After TR4 knockdown in JS-1 cells, (a) mRNA expressions of RARβ and RXRα detected by qPCR; (b) protein expressions of RARβ and RXRα detected by Western blot; (c) quantification of RARβ and RXRα proteins. After lentivirus-mediated overexpression of TR4 in JS-1 cells, (d) mRNA expressions of RARβ and RXRα detected by qPCR; (e) protein expressions of RARβ and RXRα detected by Western blot; (f) quantification of RARβ and RXRα proteins. Data are presented as mean ± SD. *P<0.05, ⁎⁎P<0.01. WT, the without transfection cells; LV-control, the control lentiviral cells; shTR4, the TR4 knockdown cells; OETR4, the TR4 overexpression cells.
TR4 is a nuclear transcription factor encoded by the nuclear receptor superfamily 2, group C, member 2 (NR2C2) gene, and belongs to the nuclear receptor superfamily. Studies have shown 98% overall homology between human and rat TR4. TR4 was shown to be abundantly expressed in the hypothalamus of rats and was also detected in the liver [10]. In addition, the nuclear receptor signaling atlas in mouse liver showed that TR4 is a moderately abundant nuclear receptor in the liver [11]. However, there is a paucity of studies on the role of TR4 in liver diseases; in addition, the relationship between TR4 and liver fibrosis is not well characterized in contemporary literature.
In this study, we observed a significant increase in the expression of TR4 in BDL-induced rat fibrotic liver, accompanied by increased expression of α-SMA. These findings suggested the potential involvement of TR4 in the process of liver fibrosis and its close relation with the activation of HSCs.
TR4 is closely related to lipid metabolism. On the one hand, fatty acids can activate TR4 to regulate the transcriptional expression of the target genes, such as polyunsaturated fatty acids and their metabolites [5,12]. On the other hand, TR4 has been shown to promote fatty acid metabolism. Overexpression of TR4 was shown to promote lipid accumulation in 3T3-L1 adipocytes by inducing fatty acid uptake and synthesis [13]. In addition, loss of TR4 reduced lipid accumulation in the liver and adipose tissues [14,15]. HSCs are a kind of lipid storage cells characterized by the perinuclear distribution of a large number of lipid droplets containing vitamin A; these lipid droplets containing vitamin A were decreased or even disappeared in activated HSCs [16]. BDL-induced fibrotic liver tissues showed abnormal proliferation and activation of HSCs [8], along with a significant reduction in intracellular retinoic acid and lipid droplets; however, the expression of TR4 was increased, suggesting that the change in TR4 expression was not the result of HSCs activation. Based on this phenomenon, we speculated that TR4 might be a factor that triggered the activation of HSCs. To verify this speculation, we constructed shTR4 and OETR4 JS-1 cells via lentiviral transfection to further observe the effect of TR4 on the activation of HSCs. As speculated, TR4 knockdown significantly inhibited the activation of JS1 cells, with significantly reduced expressions of α-SMA and Col-I. On the contrary, TR4 gene overexpression promoted the activation of JS1 cells, which resulted in a significant increase in the expressions of α-SMA and Col-I. The above results suggested that TR4 triggered the activation of HSCs. Activation of HSCs has been shown to be a central event in the progression of liver fibrosis, and activated HSCs are the main source of myofibroblasts in the liver, which generate extracellular matrix and lead to the continuous progression of liver fibrosis [17]. Therefore, our results suggest that TR4 may play an important role in the progression of liver fibrosis.
Studies have reported that TR4 can regulate TGF-β1/smads signaling pathway. High expression of TR4 has been demonstrated in the CD133+ stem/progenitor cells of prostate cancer (PCa CD133+ S/P). Targeting TR4 with lentiviral silencing RNA significantly inhibited the invasive ability of PCa CD133+ S/P cells, and the underlying mechanism may be related to the regulation of TGF-β1 signaling pathway [7]. In addition, TR4 was also shown to activate the TβR2/p-Smad3 signaling pathway by inhibiting miR-373-3p expression, thereby promoting the invasion and metastasis of prostate cancer6. TGF-β1/smads signaling pathway plays a key role in the activation of HSCs and the development of liver fibrosis [18]. The binding of TGF-β1 to TGF-β1 receptor (TβR) on HSCs membrane was found to activate TβR, subsequently leading to phosphorylation and activation of Smad2/3 (a specific signal transduction protein of TβRI receptor) bound to TβR. Phosphorylated Smad2/3 formed multimeric complexes with Smad4, crossed the nuclear membrane and entered the nucleus, where they participated in the transcription of different target genes in consort with other transcription factors. Binding of Smad7 to TβRI prevented the activation of receptor-activated smads (R-smads), thereby inhibiting the TGF-β1 signal transduction mediated by the activation of R-smads [19]. In this study, TR4 knockdown significantly reduced the protein expressions of TGF-β1, p-TβRI, and p-smad2/3 in JS1 cells, and TR4 overexpression significantly increased the expressions of p-TβRI and p-smad2/3. These results suggested that TR4 enhanced the transduction of TGF-β1/smads signaling pathway by increasing the expression level of p-TβRI and p-smad2/3, thereby promoting the activation of HSCs. Of note, after TR4 overexpression, there was no significant change in the expression of TGF-β1 in JS1 cells. The reason may be related to the activation state of JS1 cells. Since the JS1 cells were activated, and TGF-β1 was already at a high level, further increasing its expression was not easy. Although TGF-β1was not increased significantly after TR4 overexpression, the protein expressions of p-TβRI and p-smad2/3 were still significantly increased, suggesting that TR4 promoted the transduction of TGF-β1/smads signaling pathway, which may not be completely dependent on the activation of TGF-β1. These findings are similar to those reported by Qiu et al. [6], who found that TR4 promoted the invasion and metastasis of prostate cancer cells by increasing the expression of TβR2 and p-smad3, independent of TGF-β1 itself. Based on the above results, our findings suggest that TR4 promoted the activation of HSCs by up-regulating the TβRI/Smad2/3 signaling pathway.
In addition, TR4 was shown to be a vitamin A-activated nuclear receptor as a negative feedback regulator of the retinoic acid-retinol receptor signaling pathways [20,21], which were closely related to HSCs. We wondered whether TR4 also played a role in inhibiting the activation of HSCs by regulating this pathway. As shown in this research, TR4 knockdown induced a significant increase in the expression of RXRα, while TR4 overexpression had the opposite effect. However, the above conditions had no significant effect on RARβ signaling, which suggested that the reduction of RXRα expression in HSCs by TR4 may be part of its mechanism of promoting HSCs activation; however, further research is required to corroborate this speculation.
5ConclusionsIn conclusion, to the best of our knowledge, this is the first study to demonstrate the potential role of TR4, an important nuclear transcription factor, in the activation of HSCs. TR4 triggered HSCs activation by up-regulating TβRI/Smad2/3 signaling pathway and reducing the level of RXR α, thereby participating in the progression of liver fibrosis (Fig. 6). Our findings suggest that targeting TR4 may be a novel anti-fibrosis strategy in the future.
Author's contributions
Yadong Fu, Yuping Zhou, Ping Liu and Jiamei Chen had substantial contributions to the conception and design of the manuscript; Yadong Fu, Yuping Zhou, Yongping Mu, Ying Lv, Gaofeng Chen, Hua Zhang provided materials, performed the experiments, collected the data, and analyzed the results. Yadong Fu, Ping Liu and Jiamei Chen assisted in the writing of the article and review of intellectual content. Ping Liu and Jiamei Chen contributed to the final approval of the version to be submitted, and all authors are in agreement with all aspects of the paper.
Declaration of interestNone.
FundingThis work was supported by the National Natural Science Foundation of China (grant numbers 82130120, 81973613, 81530101) and the Shanghai Rising-Star Program (grant number 19QA1408900).



