



The increasing prevalence of obesity in Western countries has led to a significant increase of nonalcoholic fatty liver disease (NAFLD) over the past decades. Being part of the metabolic syndrome, NAFLD is thought to be the most frequent cause of elevated liver enzymes in the United States affecting up to one third of the population. NAFLD is also proposed to be the major cause for cryptogenic cirrhosis and hepatocellular cancer of unknown etiology, and thus, represents one of the most important problems for hepatologists in the future. However, the natural course of NAFLD is highly variable and is influenced by both environmental and genetic factors. Polymorphisms in specific genes have been proposed to increase the risk of fibrosis in patients with NAFLD. The present review article summarizes currently available data from genotype-pheno-type studies and defines candidate genes that deserve future investigation.
Abbreviations
Nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), hepatocellular cancer (HCC), tumor necrosis factor alpha (TNF-α), free fatty acids (FFA), reactive oxygen species (ROS), triglycerides (TG), microsomal triglyceride transfer protein (MTP), very low density lipoprotein (VLDL), alanine aminotransferase (ALT), phosphatidylethanolamine N-methyltransferase (PEMT), carnitine palmitoyl-transferase 1A (CPT-1A), manganese-dependent superoxide dismutase (MnSOD), hereditary hemochromatosis (HHC), glutathione-s-transferase (GST), myeloperoxidase (MPO), microsomal epoxide hydrolase (MEH), lipopolysaccharide (LPS), Toll-like receptor-4 (TLR4), hepatic stellate cells (HSC), platelet derived growth factor (PDGF), matrix metalloproteinases (MMP), primary sclerosing cholangitis (PSC), nonalcoholic steatohepatitis (NASH), methylene tetrahydrofolat reductase (MTHFR), extracellular matrix (ECM).
IntroductionThe epidemic of obesity in the Western has increased the prevalence of nonalcoholic fatty liver disease (NAFLD) rendering it now the leading cause of referral to hepatology clinics. NAFLD represents the hepatic manifestation of the metabolic syndrome which is characterized by obesity, type 2 diabetes mellitus, and dyslipidemia, with insulin resistance as a common feature. Indeed, it is estimated that 17-33% of Americans are affected. NAFLD is now believed to be the major cause for abnormal liver function and fibrosis in industrialized countries.1 NAFLD refers to a spectrum of histological findings ranging from simple and reversible steatosis to steatohepatitis and cirrhosis, and is diagnosed after ruling out other causes, in particular alcoholic liver disease (ALD).2 NAFLD may account for the majority of cryptogenic cirrhosis cases. In this respect, features suggestive of NAFLD such as obesity or insulin resistance are more frequently observed in patients with cryptogenic cirrhosis than in age-and gender-matched patients with cirrhosis of known etiology. Remarkably, hypertriglyceridemia and diabetes are independent risk factors for the development of hepatocellular cancer (HCC) in patients with cryptogenic cirrhosis, suggesting that HCC represents a complication in an unknown percentage of cases of nonalcoholic steatohepatitis (NASH) induced cirrhosis.3
While the majority of obese people only develop simple hepatic steatosis, around 10–20% of morbidly obese patients progress to more advanced disease.4,5 The current concept of liver fibrosis suggests that the natural history of liver diseases in general is influenced by both environmental and genetic factors that act in concert.6 The former are not sufficient to explain the wide variety of phenotypes among individuals subjected to profibrogenic insults, and the latter are largely unknown. While the risk factors for NAFLD are well established, little is known about genetic factors.
Fibrosis in NAFLD is thought to be the result of a nonspecific wound-healing response to inflammation and repeated injury, ultimately resulting in the formation of scar tissue instead of parenchyma.6 Given the histological similarity between NAFLD and ALD it is not surprising that an emerging body of evidence suggests that their pathological mechanisms are very similar and involve cytokine and oxidative stress-mediated injury. Tumor necrosis factor alpha (TNF-α) is of special interest in the pathogenesis of NAFLD and may be produced by Kupffer cells in response to gut derived endotoxin, but also by hepatocytes in response to an increased supply of free fatty acids (FFA), or by adipose tissue macrophages.2,7 Reactive oxygen species (ROS) in the setting of NAFLD principally arises via increased oxidation of FFA by mitochondria, peroxisomes, and microsomes. In addition, there is emerging evidence that factors related to obesity and insulin resistance may be directly fibrogenic.7 These include insulin and adipokines synthesized and released by adipocytes, including angiotensinogen, norepinephrine, and leptin. The anti-inflammatory and anti-steatotic adipokine adiponectin may also contribute to steatosis and inflammation in NAFLD and protects from NAFLD associated carcinogenesis as suggested by studies carried out in mice8 (Kamada Y, Journal of Hepatology, in press).
Evidence for genetic factorsThere is convincing evidence that genetic factors account for considerable variability in the natural history of NAFLD. Evidence for a contributing effect of genetic factors on the development of advanced fibrosis in NAFLD derive from family clustering studies showing that about one fifth of patients with NASH have a similarly affected first degree relative.9 In another study, coexistence of NASH and cryptogenic cirrhosis was observed in seven out of eight families studied.10 In addition there appears to be interethnic variations in susceptibility towards NAFLD. In this respect, it has been reported that the prevalence of cryptogenic cirrhosis in Hispanic and African-Americans was threefold higher and fourfold lower, respectively, compared with Americans of European origin. Assuming that these groups share a similar prevalence of type 2 diabetes, this observation suggests that Hispanics are at particularly high risk.11
Compared to chronic HCV infection and ALD,2,13 only little is known about factors influencing fibrosis progression in patients with NAFLD. With regard to the pathomechamisms underlying NAFLD, variations in genes affecting hepatic lipid metabolism, insulin resistance, ROS formation and degradation, cytokines and endotoxin receptors, and profibrogenic mediators are of special interest.
Genes influencing lipid metabolism and steatosisStudies from Japan suggest that genetic predisposition to obesity and inflammation contributes to the development of NASH.4,15 These studies investigated functional genetic variants of β-adrenergic receptors as they play an important role in the regulation of energy expenditure, in part, by stimulating lipid mobilization through lipolysis. Patients with NASH carried significantly more frequently the codon 64 Arginine variant of the β3-adrenergic receptor than control subjects, which results in hypertriglyceridemia, hyperinsulinemia and obesity.14 Homozygous carriers of Glycine at codon 16 of the β2 adrenergic receptor have lower high-density lipoprotein cholesterol level than Arginine homozygotes, and presence of Glutamine at codon 27 is associated with higher concentrations of serum triglycerides (TG) and a higher prevalence of steatosis.15
The microsomal triglyceride transfer protein (MTP) regulates synthesis, storage, and export of hepatic TG content and is critical for the synthesis and secretion of very low density lipoprotein (VLDL) in the liver. Low levels of MTP result in failure to excrete triacylglycerol from the liver and hepatic steatosis. Two studies provide evidence that genetic variation of the MTP gene affects susceptibility to development of NAFLD. In patients with type II diabetes, the -493 G/T MTP gene polymorphism was independently associated with elevated an serum alanine aminotransferase (ALT) level as a surrogate marker for steatohepatitis.16 Accordingly, patients with NASH had a higher frequency of the G allele and the G/G genotype compared to controls. Interestingly, the G/G genotype was also associated with more severe steatosis and more advanced stage of NASH compared to patients with genotype G/T.17 Noteworthy, the same polymorphism was also reported to affect progression to advanced fibrosis in patients with chronic HCV infection.18
A loss-of-function mutation of the phosphatidylethanolamine N-methyltransferase (PEMT) gene, which is required for phosphatidylcholine synthesis, interferes with efficient VLDL synthesis. This mutation was reported to confer susceptibility to NAFLD, and is supported by data from PEMT knock out mice which also develop fatty liver disease.19
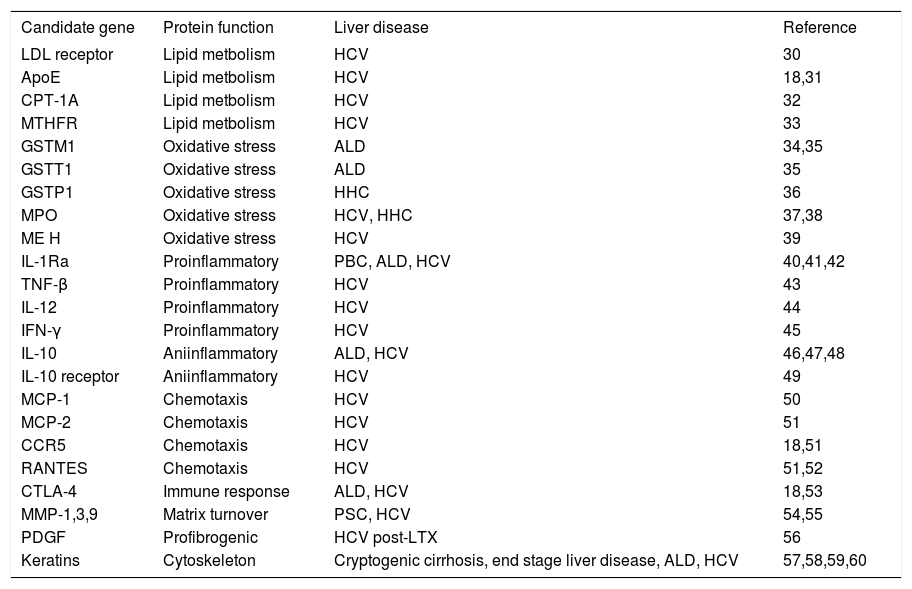
The genes coding for the LDL receptor, the ApoE protein, the methylene tetrahydrofolat reductase (MTHFR) and the carnitine palmitoyltransferase 1A (CPT-1A) also display variations that have been proven to affect the course of chronic HCV infection and thus deserve future investigation in patients with NAFLD (seetable II).
Candidate genes for future studies.
| Candidate gene | Protein function | Liver disease | Reference |
|---|---|---|---|
| LDL receptor | Lipid metbolism | HCV | 30 |
| ApoE | Lipid metbolism | HCV | 18,31 |
| CPT-1A | Lipid metbolism | HCV | 32 |
| MTHFR | Lipid metbolism | HCV | 33 |
| GSTM1 | Oxidative stress | ALD | 34,35 |
| GSTT1 | Oxidative stress | ALD | 35 |
| GSTP1 | Oxidative stress | HHC | 36 |
| MPO | Oxidative stress | HCV, HHC | 37,38 |
| ME H | Oxidative stress | HCV | 39 |
| IL-1Ra | Proinflammatory | PBC, ALD, HCV | 40,41,42 |
| TNF-β | Proinflammatory | HCV | 43 |
| IL-12 | Proinflammatory | HCV | 44 |
| IFN-γ | Proinflammatory | HCV | 45 |
| IL-10 | Aniinflammatory | ALD, HCV | 46,47,48 |
| IL-10 receptor | Aniinflammatory | HCV | 49 |
| MCP-1 | Chemotaxis | HCV | 50 |
| MCP-2 | Chemotaxis | HCV | 51 |
| CCR5 | Chemotaxis | HCV | 18,51 |
| RANTES | Chemotaxis | HCV | 51,52 |
| CTLA-4 | Immune response | ALD, HCV | 18,53 |
| MMP-1,3,9 | Matrix turnover | PSC, HCV | 54,55 |
| PDGF | Profibrogenic | HCV post-LTX | 56 |
| Keratins | Cytoskeleton | Cryptogenic cirrhosis, end stage liver disease, ALD, HCV | 57,58,59,60 |
Genetic variations of enzymes involved in generation and degradation of ROS are of special interest with regards to NAFLD, as excessive FFA oxidation leads to oxidative stress causing hepatocyte apoptosis and liver injury. Manganese-dependent superoxide dismutase (Mn-SOD) is the main ROS scavenger in mitochondria. A Japanese study has reported that the T/T genotype of the MnSOD gene which leads to less efficient transport of MnSOD to the mitochondria is a susceptibility allele for the development of NAFLD.17
The HFE gene is another attractive candidate to be studied as genetic modifier of NAFLD. Mutations of the HFE gene cause hereditary hemochromatosis (HHC), which is the most common genetic disease in populations of European ancestry. In addition, HFE gene mutations are common among Caucasians and have been demonstrated to be functional in terms of increased duodenal iron absorption and liver iron deposition. Increased liver iron can act as a catalyst promoting oxidative stress in a steatotic liver. As steatosis has been reported to affect the natural course of HHC20
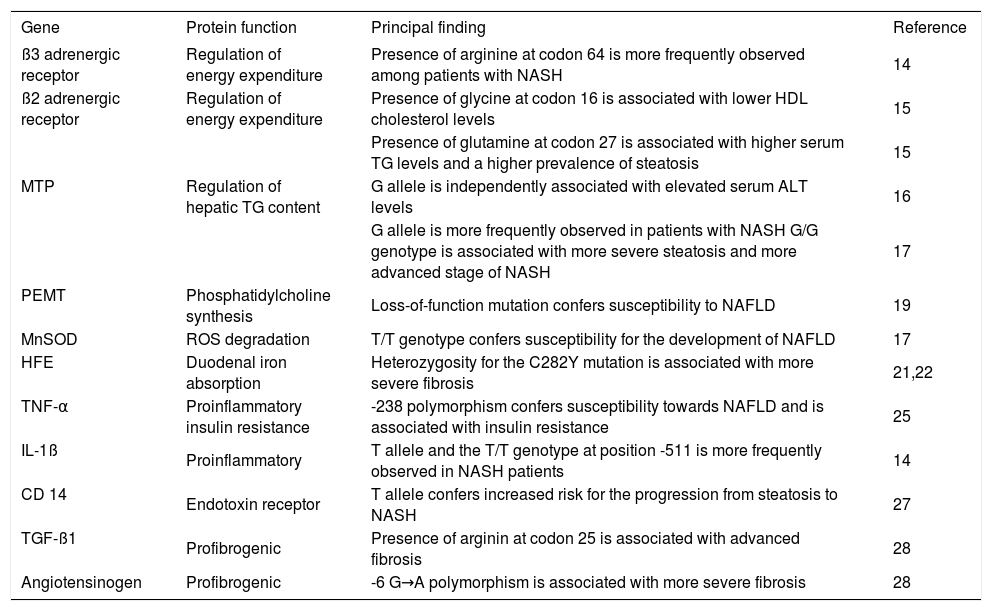
Genotype-phenotype studies in NAFLD.
| Gene | Protein function | Principal finding | Reference |
|---|---|---|---|
| ß3 adrenergic receptor | Regulation of energy expenditure | Presence of arginine at codon 64 is more frequently observed among patients with NASH | 14 |
| ß2 adrenergic receptor | Regulation of energy expenditure | Presence of glycine at codon 16 is associated with lower HDL cholesterol levels | 15 |
| Presence of glutamine at codon 27 is associated with higher serum TG levels and a higher prevalence of steatosis | 15 | ||
| MTP | Regulation of hepatic TG content | G allele is independently associated with elevated serum ALT levels | 16 |
| G allele is more frequently observed in patients with NASH G/G genotype is associated with more severe steatosis and more advanced stage of NASH | 17 | ||
| PEMT | Phosphatidylcholine synthesis | Loss-of-function mutation confers susceptibility to NAFLD | 19 |
| MnSOD | ROS degradation | T/T genotype confers susceptibility for the development of NAFLD | 17 |
| HFE | Duodenal iron absorption | Heterozygosity for the C282Y mutation is associated with more severe fibrosis | 21,22 |
| TNF-α | Proinflammatory insulin resistance | -238 polymorphism confers susceptibility towards NAFLD and is associated with insulin resistance | 25 |
| IL-1ß | Proinflammatory | T allele and the T/T genotype at position -511 is more frequently observed in NASH patients | 14 |
| CD 14 | Endotoxin receptor | T allele confers increased risk for the progression from steatosis to NASH | 27 |
| TGF-ß1 | Profibrogenic | Presence of arginin at codon 25 is associated with advanced fibrosis | 28 |
| Angiotensinogen | Profibrogenic | -6 G→A polymorphism is associated with more severe fibrosis | 28 |
A number of studies have provided evidence that polymorphisms of other genes involved in the generation or degradation of ROS such as glutathione-s-transferase-isoforms (GSTM1, GSTT1, GSTP1), myeloperoxidase (MPO) or microsomal epoxide hydrolase (MEH) affect progression to cirrhosis in different liver disease such as ALD, chronic HCV infection or HHC. Given the key role of FFA oxidation and ROS generation in the course of hepatocyte death in NAFLD these polymorphisms should be investigated in the future (seetable II).
Cytokine gene polymorphismsTNF-α plays a major role in the pathogenesis of NAFLD, as increased TNF-α levels favor the development of insulin resistance and impaired glucose tolerance. A promoter polymorphism of the TNF-α gene associated with increased cytokine expression was suggested to affect susceptibility towards NAFLD and to be associated with higher insulin resistance indices and a higher prevalence of impaired glucose tolerance.25 A Japanese study reported that the frequencies of the IL-β -511 T allele and the T/T genotype are significantly higher in NASH patients than in controls providing further evidence for the involvement of cytokine gene polymorphism in the course of NAFLD.14
Chronic inflammation and a mixed cellular infiltrate are key characteristics of NASH. It is therefore surprising that TNF-α and IL-1β are the only cytokine gene polymorphisms that have been evaluated in NAFLD so far.Table II summarizes data from studies focusing on the role of genetic variants of cytokines, chemokines, their receptors and other proteins involved in immune responses in other liver diseases that might also play a role in the progression from steatosis to NASH and more advanced stages of NAFLD.
Genes coding for bacterial receptorsThere is evidence that gut derived endotoxin leading to cytokine release contributes to the pathogenesis of NAFLD. CD14 is a co-receptor for gram negative bacterial lipopolysaccharide (LPS) expressed on monocytes, macrophages but also on other cells. CD14 lacks an intra-cellular domain but enhances signaling through Toll-like receptor-4 (TLR4). Expression of CD14 has been suggested to be influenced by a polymorphism within the promoter region. Similar to patients with ALD,26 the T al-lele was suggested to confer increased risk for the progression from steatosis to NASH.27 The Asp299Gly polymorphism in the TLR4 gene has been associated with endotoxin hyporesponsiveness in humans. This polymorphism definitely deserves to be tested in larger cohorts of patients with NAFLD.
Genes influencing extracellular matrix synthesis and degradationHepatic stellate cells (HSC) are believed to be the major collagen-producing cells in liver fibrosis. Upon activation by cytokines and other agonists, HSC acquire a profibrogenic phenotype and synthesize extracellular matrix ECM that replaces parenchymal tissue. Consequently, variations of genes that play a crucial role in the activation of HSCs should affect progression to cirrhosis irrespective of etiology and the underlying cause of liver disease. One would expect that variations of these genes affect progression to cirrhosis in a uniform and global way and are therefore primary candidates to be studied in NAFLD-related fibrosis. The only relevant study so far in this regard in NAFLD reported that the combination of high angiotensinogen and TGF-β1 producing polymorphisms is associated with advanced hepatic fibrosis in obese patients with NAFLD28 which is in accordance with findings in patients with chronic HCV infection.29
Genetic variation of the platelet derived growth factor (PDGF), a key molecule in liver fibrosis driving HSC proliferation, has been reported to affect the course of HCV infection after liver transplantation. So far data is lacking if this polymorphism also contributes to the course of other chronic liver diseases including NAFLD. Matrix metalloproteinases (MMP) constitute another group of attractive candidate genes to be studied in NAFLD as they have been shown to affect the course of HCV virus infection and primary sclerosing cholangitis (PSC) by interfering with adequate matrix turn-over. Mallory body formation is a characteristic histopathological feature of NASH that results from misfolding and aggregation of keratins. There is convincing evidence that mutations of keratins can cause cryptogenic cirrhosis and affect the course of liver diseases of different etiology (seetable II). Results from studies investigating the role of keratin mutations in NAFLD are anticipated with great interest.
ConclusionNAFLD has become the leading cause of referral to hepatology clinics and a major future challenge. Data from family studies and interethnic differences in susceptibility suggest that genetic factors contribute to the variability seen in natural course of NAFLD. Identifying risk genes that predict faster fibrosis progression or susceptibility for HCC development might help managing patients in an individual way. However, data from gene association studies carried out in patients with NAFLD are inconclusive. Most published studies were carried out in either small cohorts of patients carrying the risk of type I error or with inaccurate study design. Available reports need to be verified by subsequent larger studies. A number of other candidate genes that have been suggested to contribute to fibrosis progression in other chronic liver diseases have not been evaluated. Carefully designed studies according to recently defined guidelines13 requiring collaboration of different research centers will help to understand the role of genetics of NAFLD.
AcknowledgementsChristoph H. Österreicher is supported by an Erwin Schroedinger research fellowship kindly provided by the Austrian Science Fund (FWF). David A. Brenner is supported by grants from the NIH.



