



Oxidative stress is a major pathogenetic event occurring in several liver disorders ranging from metabolic to proliferative ones, and is a major cause of liver damage due to Ischemia/Reperfusion (I/R) during liver transplantation. The main sources of ROS are represented by mitochondria and cytocrome P450 enzymes in the hepatocyte, by Kupffer cells and by neutrophils. Cells are provided with efficient molecular strategies to strictly control the intracellular ROS level and to maintain the balance between oxidant and antioxidant molecules. A cellular oxidative stress condition is determined by an imbalance between the generation of ROS and the antioxidant defense capacity of the cell and can affect major cellular components including lipids, proteins and DNA. Proteins are very important signposts of cellular redox status and through their structure/function modulation, ROS can also influence gene expression profile by affecting intracellular signal transduction pathways.
While several enzymatic (such as superoxide dismutase, catalase, glutathione peroxidase) and non enzymatic (such as 4-hydroxynonenal, decrease of glutathione, vitamin E, vitamin C, malondialdehyde) markers of chronic oxidative stress in liver are well known, early protein targets of oxidative injury are yet not well defined. Identification of these markers will enable early detection of liver diseases and will allow monitoring the degree of liver damage, the response to pharmacological therapies and the development of new therapeutic approaches. In the new era of molecular medicine, new proteomics methodologies promise to establish a relationship between pathological hallmarks of disease and protein structural and functional abnormalities in liver disease, thus allowing a better understanding and a more rational therapy on these disorders.
Abbrivations
APE/Ref-1, Apurinic Apyrimidinic Endonuclease/Redox Effector Factor-1; AP-1,Activator Protein-1; CAT, catalase; GSH, glutathione; GPx, glutathione peroxidase; I/R, Ischemia/Reperfusion; LT, Liver Transplantation; NF-kB, Nuclear Factor-k B; ROS, Reactive Oxygen Species; SOD, superoxide dismutase
IntroductionI.a. General considerations on cellular redox state.Reactive oxygen species (such as H2O2, OH., O2., collectively known as ROS) play important physiological functions and can also cause extensive cellular damage. Cells are provided with efficient molecular strategies to strictly control the intracellular ROS level and to maintain the balance between oxidant and antioxidant molecules. Oxidative stress, resulting from an imbalance between the generation of ROS and the antioxidant defense capacity of the cell,1 affects major cellular components, including lipids, proteins, and DNA. This phenomenon is closely associated with a number of human disorders such as many degenerative diseases, including cardiovascular disease, diabetes, cancer and neurodegenerative disorders2,3 and with almost all liver pathologies.4-6 All these conditions appear to be mostly related to chronic oxidative stress. However, the acute exposure to high levels of ROS seems also to be responsible for the development of different damage such as during ischemia/reperfusion (I/R) in liver.7,8
Besides to produce cell damage, ROS can also be considered as molecular second messengers within the cell as they can be generated during triggering of particular cellular responses by cytokines, hormones, growth factors and other soluble mediators such as extracellular ATP.2
Therefore, ROS represent a double face medal and could act either positively or negatively on cell functioning depending on the intensity and duration of the oxidative stress produced on a cell. It is therefore not surprising the role of ROS either as apoptotic molecules or stimulators of cell proliferation, depending on the cell type and on the intensity of the stress produced.
I.b. Major types of oxygen free radicals and their derivativesMitochondria are the major source of cellular ROS in non-phagocytic cells.
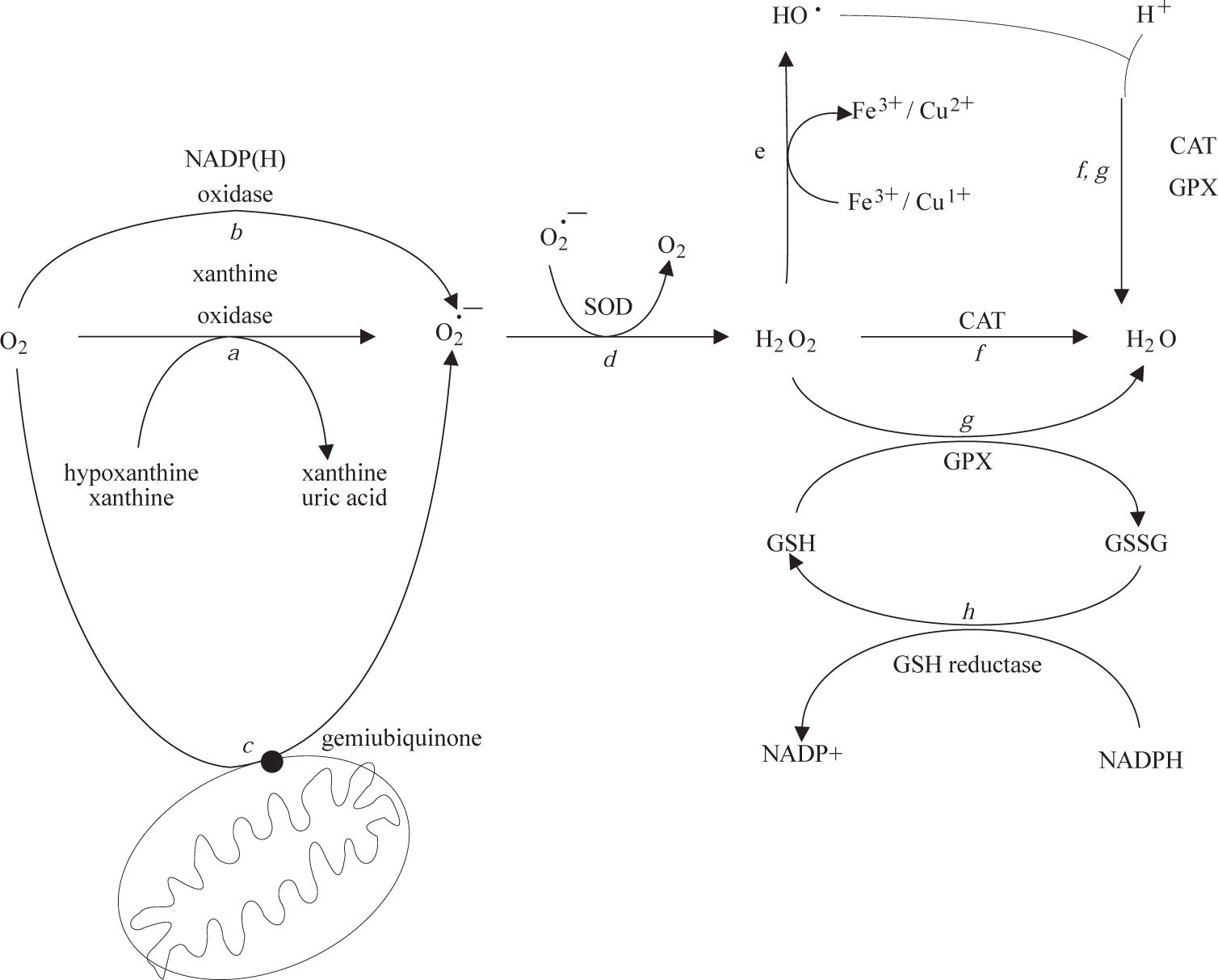
Low levels of potentially toxic oxygen metabolites are physiologically generated in cells and tissues during oxidative phosphorylation.10 The resulting moderate levels of ROS play an integral role in the modulation of several physiological functions of the cell, including gene expression, signal transduction, and defense against invading pathogens. Under normal conditions, it is estimated that up to 1% of the mitochondrial electron flow primarily leads to the formation of superoxide anion that is formed by the univalent reduction of molecular oxygen. This process is mediated by enzymes such as NADP(H) oxidases and xanthine oxidase (Figure 1, reactions a and b) or nonenzymatically by redox-reactive compounds, such as the semi-ubiquinone compound of the mitochondrial electron transport chain (Figure 1, reaction c). Interference with electron transport can dramatically increase superoxide production. Superoxide is rapidly converted within the cell to H2O2 and O2 by the superoxide dismutases (SODs)11(Figure 1, reaction d). H2O2 can react with reduced transition metals, via the Fenton reaction, to produce the highly reactive hydroxyl radical (Figure 1, reaction e), a far more damaging molecule to the cell. Alternatively, H2O2 may be converted into water by the enzymes catalase and glutathione peroxidase (Figure 1, reactions f and g). During the glutathione peroxidase reaction, glutathione is oxidized to glutathione disulfide, which can be the reconverted to glutathione by glutathione reductase in an NADPH-consuming process (Figure 1, reaction h).
. SOD, superoxide dismutase; CAT, catalase; GPx, glutathione peroxidase.")
Frequently, different reactive oxygen species coexist in the reactive cellular environment resulting in a difficult identification of the real effector molecule responsible for a given biological effect.
Oxidative stress may cause reversible and/or irreversible modifications on sensitive proteins. Reversible modifications, usually at Cysteine residues, may have a dual role: i) protection from irreversible damage and ii) modulation of protein function (redox regulation). Irreversible modifications induced by ROS, such di-Tyrosine formation, protein-protein cross-linking or Lysine and Arginine carbonylation, are generally associated with permanent loss of function and may lead to either the degradation of the damaged proteins or their progressive accumulation into cytoplasmic inclusions, as observed in age-related neurodegenerative disorders.3
I.c. Oxidative stress response as a model of redox signallingRedox signalling implies the use of redox chemistry in controlling cellular responses through signal transduction mechanisms. Redox signalling is a highly conserved biological mechanism during phylogenesis of living organisms. Interestingly, it resulted in a widely used molecular strategy by different organisms (ranging from bacterial procariotes to higher eukaryotes) probably as a consequence of the evolutive pressure represented by the abundance of oxygen in the Hearth atmosphere. ROS are present in cells and tissues at low but measurable concentration which is determined by the balance between the rate of production versus the clearance by different antioxidant enzymes (SOD, glutathione peroxidase and catalase) and compounds (glutathione, ascorbate, α-tocopherol, β-carotene).12
A complex functional modulation of several proteins occurs upon cellular oxidative stress.13-15 In particular, this is accomplished through modification of proteins since several aminoacids, such as Tryptophan, Tyrosine, Histidine and particularly Cysteine, are direct targets of ROS.16 ROS-mediated protein modification can affect both protein structure and function and protein stability. It is largely known that oxidized proteins are highly sensitive to proteolytic attack by proteosomes.17 By controlling the functional activity of cellular effectors (i.e. proteins) it is clear that ROS can also affect signal transduction mechanisms and eventually, influence cellular gene expression. It is becoming increasingly evident that mammalian cells can modulate patterns of protein expression in response to hydroperoxide stress, by activating redox-sensitive transcription factors such as Egr-1, NF- κB and AP-1, G-proteins and by activating cellular kinases such as the mitogen-activated protein kinase (MAPK) family.18 This oxidative-dependent cellular signalling pathways alteration may frequently lead, with a still unknown mechanism, to hepatocyte apoptosis. However, sensor polypeptides specific to H2O2 and other ROS are still to be identified and little is known about early effects on biological macromolecules regulating ROS-induced cellular effects. It is clear that the understanding of stimulus-specific mechanisms of oxidant-dependent hepatocyte apoptosis would be extremely important to develop effective therapies for a number of forms of liver injuries.
I.d. Redox sensitive targets in hepatocytes: the central role of APE/Ref-1 as a molecular paradigmIt is well established that elevated H2O2 levels are produced in different liver disorders4,19 as well as during ethanol methabolism.20,21 The normal liver is provided with very efficient enzymatic and non enzymatic antioxidant systems.22 In particular, Kupffer cells and hepatic stellate cells are potentially more exposed to ROS molecules and it has been well documented that hepatic antioxidant systems are significantly decreased in several chronic liver diseases.22,23 Although the study of the involvement of the classical intracellular ROS molecular scavengers, such as SOD, CAT, GPx, is of fundamental importance in setting up therapeutical approaches toward oxidative-based liver pathologies, understanding of the molecular switches involved in cell oxidative stress response at the genomic level could also provide interesting applications, in particular the identification of the early ‘molecular switches’ of the redoxbased cellular response. This implies that a more focused approach should be reserved to understanding those mechanisms through which cells may change their genomic array upon oxidative burst shortly after injury.
Although information is still scanty, some genes and their protein products have been discovered to be of fundamental importance in controlling the cell function during oxidative stress conditions by acting at the genomic level and not simply as ROS scavengers. APE/Ref-1 is a paradigmatical example of this mechanism and provides a good example of the biological complexity of the system.24 APE/Ref-1 protein plays a central role in ROS-induced cellular effects, as a major member of the base excision repair (BER) pathway of DNA repair, on apurinic/apyrimidinic (AP) sites in DNA (generated as a consequence of oxidative-induced damage on DNA) and as redox controller of the activity of several important transcription factors (TFs) such as NF-κB, AP-1, HIF-1α and Pax proteins. APE/Ref-1 has been shown to inhibit apoptosis and its altered level or cellular location have been described in several human pathologies. Therefore, APE/Ref-1 appears to form a unique link between the DNA BER pathway, oxidative signaling and cellular injury. APE/Ref-1 protein contains two functionally distinct domains. The N-terminal domain contains the nuclear localization sequence and is essential for redox activity, while the endonuclease activity resides in the C-terminal region. APE/Ref-1 is regulated at both the transcriptional and post-translational level. In terms of transcriptional regulation, the effects of ROS on APE/Ref-1 induction have been the most intensely studied. Data reported so far demonstrated that oxidative agents, such as H2O2, and ROS-generating injuries such as UV-radiation, promote a transient APE/Ref-1 protein and mRNA induction which correlates with an increase of its endonuclease and redox activities. At present, it is not known both what is the ‘primum movens’ responsible for APE1/Ref-1 gene expression upon oxidative stress and the signalling pathways leading to its activation. Since APE1/Ref-1 expression represents a molecular marker of oxidative stress, it would be important to understand the dissection of signaling pathways responsible for its role in regulating the mechanisms of ROS-induced cell responses. The posttranslational regulation of APE/Ref-1 activities seems to reside into two non-mutually exclusive mechanisms, i.e. subcellular localization and phosphorylation degree. On one side, APE/Ref-1 undergoes an active cytoplasm to nucleus translocation in different cells25,26 upon ROS exposure. On the other side, phosphorylation and acetylation seem to play a role in determining the functional activity of the protein, at least ‘in vitro’. However, neither molecular mechanisms responsible for the APE/Ref-1 gene expression induction upon oxidative injury nor functional data regarding the real ‘in vivo’ role played by post-translational modifications in controlling APE/Ref-1 functional activities are clear.24 Understanding the possible relationships existing between APE/Ref-1 levels and oxidative-stress-based human liver pathologies is of enormous importance, not only for the comprehension of the role and mechanism that APE/Ref-1 may play in the initiation and development of these pathologies but also for developing diagnostic markers and therapeutic tools.
I.e. Pathophysiological implications of redox regulation in liverROS play a crucial role in the induction and progression of different liver diseases and evidence of oxidative stress has been detected in almost all the clinical and experimental conditions of chronic liver diseases with different etiology and progression rate of fibrosis.4,5,27 The pathogenesis of the damage involves all the cell types present in the liver (hepatocytes, Küppfer, stellate and endothelial liver cells) via apoptosis, necrosis, ischemia and regeneration, all processes leading to altered gene expression.4 The main sources of free radicals are represented by neutrophils, endotoxin-activated Küppfer cells, hepatocyte mitochondria and cytochrome P450 enzymes.6 The relevance of cellular redox imbalance in liver diseases is outlined by a number of studies in patients with viral or alcoholic liver diseases showing a correlation between liver damage and increase in pro-oxidant cellular markers such as malondialdehyde, 4-hydroxynonenal and their protein adducts, associated with a concomitant decrease of GSH, vitamin E, vitamin C, selenium, etc.4,28 These markers may contribute to monitor the degree of liver damage and the response to antiviral therapies. However, alteration of these markers only occurs in a state of chronic imbalance of cellular redox status and possibly occurs as epiphenomenon due to the particular stress the cells undergo. At present, informations about early molecular targets of oxidative stress in liver are still scanty, although they may be of the utmost interest to design innovative therapeutical approaches.
Iron overload and oxidative stressDifferent hepatocellular injuries are associated with iron overload such as inflammation, fibrosis, cirrhosis and hepatocellular carcinoma. Since iron is a well known catalyst for generation of ROS, it is not surprising that oxidative stress is a common finding in iron overload conditions [23]. Clinical relevance of this observation is reinforced by prevention of liver injury progression by antioxidant supplementation in animal models of iron overload [29].
Ethanol metabolism and oxidative stressThe oxidative metabolism of ethanol within hepatocytes by the cytochrome P450 2E1 (CYP2E1), located in the endoplasmic reticulum, has been recognized to contribute to the ethanol-induced liver damages through the generation of oxidative stress. The ethanol-inducible form of P450 2E1 has been recognized as one of the prevailing mechanisms responsible for the generation of superoxide anion from ethanol and subsequent ethanol-induced lipid peroxidation.30,31 with a major involvement of GSH depletion.32 P450 2E1 has a very efficient NADPH oxidase activity which generates substantial amount of superoxide anion and H2O2 as well as hydroxyethyl radicals, all likely to be responsible for ethanol induction of oxidative stress and lipid peroxidation.
Moreover, also a by-product of ethanol metabolism such as acetaldehyde, which is further oxidised to acetate by mitochondrial aldehyde dehydrogenase, seems to contribute primarily to the alcohol-induced liver diseases by acting at the genomic level through up-regulation of some redox-sensitive transcription factors involved in fibrogen-esis, such as AP-1 and NF-κB. This upregulation will in turn up-regulate at the transcriptional level the expression of proinflammatory cytokines (IL-1β, IL-6, IL-18, etc.), chemokines, adhesion molecules, Fas ligands33 which will activate the caspase cascades (apoptosis) and the ATP depletion from mitochondria (necrosis) leading to an autosustaining and autopotentiating oxidative loop. This is the molecular basis of the long known observation that the oxidative damage plays an important role in hepatic fibrogenesis.5
Non-alcoholic steatohepatitis (NASH), aging and oxidative stressNASH is a liver disease characterized by histopathological features similar to those of alcoholic liver disease occurring in the absence of significant alcohol consumption.34 It has been proposed that the oxidative stress may be generated by the accumulation of free fatty acids in mitochondria, saturation of mitochondrial β-oxidation and excess H2O2 production during peroxisomal β-oxidation and to CYP2E1 induction. Although of great potential important clinical implication, this conclusion needs to be further demonstrated in both experimental and human models.
A significant reduction of antioxidant defenses is usually associated with aging and leading to an increased susceptibility to oxidative stress.35 This reduction may explain, at least in part, the increased rate of fibrosis progression seen in chronic HCV patients.36
Viral hepatitis and oxidative stressCellular damage in viral hepatitis is predominantly determined by an immunological response and a large amount of data demonstrate that the persistence of infection, the progression of liver damage and, eventually, carcinogenesis are all steps in which oxidative-stress mediated pathways are involved.4 Hepatitis C virus (HCV) infection is associated with elevated levels of circulating ROS in the serum of patients.37 This increase in ROS seems to be due at least in part, to a direct activation by the HCV NS3 protein of the NADPH oxidase in monocytes38 and by the core protein and NS5A on other cells, with mechanisms involving intracellular calcium mobilization.39,40 Since chronic HCV infection may be associated with the development of hepatocellular carcinoma (HCC) and alteration of cellular redox state is also associated with tumor transformation and progression, these links could explain some aspects of the pathogenesis of hepatocarcinogenesis in HCV infection. However, since the molecular mechanisms of this potential association are far from being understood, caution is needed to extrapolate what observed in the in vitro or animal models to the much more complex human setup.
Oxidative stress during Ischemia/ReperfusionIschemia/Reperfusion (I/R) is a mandatory procedure transplantation and liver is not the exception. ROS are generated very early after reperfusion of ischemic tissue, resulting in oxidative stress that is major cause of cellular injury.8 During ischemia, the physiologic form of the Xanthine dehydrogenase is converted to the ROS-producing form Xanthine oxidase.41 On reoxygenation, Xanthine oxidase reacts with molecular oxygen to produce ROS. Therefore, the major oxidative burst during I/R occurs after the reexposure to oxygen during the so called “warm ischemia time”.7 Although the molecular mechanisms involved in I/R cellular responses are still not fully understood, a major role seems to be played by APE/Ref-1 and NF-κB proteins in liver.42 Overexpression of APE/Ref-1 by adenoviral vector resulted in suppression of the reperfusion-generated oxidative stress in a mouse model.42
Liver transplantation (LT) is at present the only available choice for the treatment of end-stage hepatic disease. The I/R damage of transplanted livers is one of the main determinants of primary non function or initial poor function of the graft conditions that may contribute to a poor transplant outcome. In addition, livers from older donors and organs with mild to moderate steatosis are more susceptible to I/R damage. Therefore, it is clear that the elucidation of the molecular mechanisms involved in the early I/R damage after liver transplantation might greatly help to develop protocols aiming at the protection of the graft thereby contributing to the improvement of the results of liver transplantation from one hand and to the broadening of the selection criteria for donor organs.
II. Present and future PerspectivesSince oxidative stress is a common pathogenetic event occurring in several liver disorders, ranging from hepatitis to cancer, the identification of a pattern of molecular alterations present at early stages of oxidative damage would be of great help in monitoring the progression of the disease and in its early recognition. To address this important target, further innovative approaches deriving from post-genomic discoveries are needed to better understand the complex molecular mechanisms of oxidative-stress cell responses and to burst the antioxidant ability of hepatocytes.
Information regarding the nature of ROS, as well as the localization and the effects of oxidative stress, may be obtained from the analysis of discrete biomarkers isolated from tissues and biological fluids. Biomarkers are cellular indicators of the physiological state and changes during a disease at a specific time. However, the presence of oxidatively damaged molecules could simply reflect secondary epiphenomena rather than having a causal role. Although a clear definition of the cause-effect role cannot be given at present, a growing body of evidence indicates that high levels of ROS induce distinct pathological consequences that greatly amplify and propagate liver injury, leading to irreversible cell and tissue degeneration.
To understand the molecular bases of oxidative-stressinduced liver diseases it is important to identify those genes whose expression/function is directly modified by oxidative stress. Modification of gene expression can be tested by two different approaches. The first consists in the study of the expression of single candidate genes already known to be target of redox-regulation, such as cjun43 and NF-κB.44 However, since it has been shown that the biological response due to oxidative stress is mediated by the modification of a complex array of genes,45 the study of single candidate genes may not be adequate to solve the complexity of oxidative-stress-based liver pathologies. The second approach consists in technologies, such as differential display, DNA microarrays or Proteomics, able to simultaneously measure modifications of a very large number of genes/gene products. At present, there is a poor knowledge on the response of liver cells upon oxidative stress in terms of protein species involved, because most studies have been carried out only at the RNA level.
The term Proteomics describes the possibility to apply global experimental procedures to evaluate gene expression in terms of single protein species characteristic of each cell type.46 Proteomic approaches compete with other experimental approaches devoted to investigate global expression profiles such as DNA microarrays but, dealing with the real effectors of biological functions, it should be able to provide with much more valuable informations. The possibility to study the early molecular effects of oxidative stress by Proteomics approach represents a powerful tool to generate new hypothesis for future studies.
By using a Proteomic approach, we recently studied the early molecular targets of oxidative stress in human epithelial lens cells, another system very susceptible to oxidative stress.47 This is one of the few examples on how a new development of Proteomics, called Redox Proteomics, may be useful in the understanding of the molecular modifications induced by oxidative stress. However, the road we just started is long and still partially obscure and it is clear that the scientific adventure has just begun. However these studies indicate the promising aspect of the Proteomics approach in the detection of early molecular markers of oxidative stress. These techniques satisfy the necessity of the identification of optimal targets for the so called ‘redox gene therapy’ that has been already demonstrated to overcome problems associated to oxidative damage during I/R after liver transplantation.48 Therefore, rather than delivering through transgene vectors, classical oxidant scavengers, such as SOD or CAT, the overexpression of early genes responsive to oxidative stress could be most efficient in protecting cells from acute doses of ROS and would provide an optimal tool to face the majority of liver pathologies. The future will tell us if this fascinating hypothesis will be clinically valuable.
III. AcknowledgementsThis work was supported by grants from MIUR (FIST 2003 and FIRB RBNE0155LB-03) to G.T.



