



The incidence of non-alcoholic fatty liver disease (NAFLD) is increasing. Previous studies indicated that Liraglutide, glucagon-like peptide-1 analogue, could regulate glucose homeostasis as a valuable treatment for Type 2 Diabetes. However, the precise effect of Liraglutide on NAFLD model in rats and the mechanism remains unknown. In this study, we investigated the molecular mechanism by which Liraglutide ameliorates hepatic steatosis in a high-fat diet (HFD)-induced rat model of NAFLD in vivo and in vitro.
Materials and methodsNALFD rat models and hepatocyte steatosis in HepG2 cells were induced by HFD and palmitate fatty acid treatment, respectively. AMPK inhibitor, Compound C was added in HepG2 cells. Autophagy-related proteins LC3, Beclin1 and Atg7, and AMPK pathway-associated proteins were evaluated by Western blot and RT-PCR.
ResultsLiraglutide enhanced autophagy as showed by the increased expression of the autophagy markers LC3, Beclin1 and Atg7 in HFD rats and HepG2 cells treated with palmitate fatty acid. In vitro, The AMPK inhibitor exhibited an inhibitory effect on Liraglutide-induced autophagy enhancement with the deceased expression of LC3, Beclin1 and Atg7. Additionally, Liraglutide treatment elevated AMPK levels and TSC1, decreased p-mTOR expression.
ConclusionsLiraglutide could upregulate autophagy to decrease lipid over-accumulation via the AMPK/mTOR pathway.
At present, non-alcoholic fatty liver disease (NAFLD) is prevalent and keeps rising. In developed countries, about 80% of adults with obesity or diabetes has been suffering from NAFLD [1,2], and the NAFLDs has relatively higher risk of liver fibrosis and liver cancer [3,4]. It is generally believed that NAFLD is closely related to insulin resistance, obesity, diabetes, dyslipidemia, and other metabolic syndromes that clinically manifest in the liver [5–7]. NAFLD includes simple fatty liver, fatty hepatitis, and fatty liver-related cirrhosis, which can develop into liver cancer [8]. NAFLD has also been considered as a high risk to induce cardiovascular and cerebrovascular diseases [9].
It is well understood that insulin resistance leads to excessive intake of free fatty acids (FFA) caused by steatosis. In addition, other factors, including oxidative stress injury, lipid peroxidation and abnormal cytokines, can cause local inflammatory changes that cause steatohepatitis in the hepatic lobules [10]. While this ‘two-hit’ hypothesis is currently widely accepted, NAFLD pathogenesis is not yet fully understood [11]. Excessive lipid deposition in the liver has been considered the primary factor that induces liver lipotoxicity [12–14]. Additionally, autophagy has been closely associated with intrahepatic lipid deposition. Previous studies have shown that autophagy degrades fat in hepatocytes as a relatively fixed process of auto-catabolism [15–17]. Autophagy can also promote “self-digestion” of accumulated, failing protein aggregates and defective organelles within the cell to maintain stability in the intracellular environment [18,19]. Lipid drops (LDs), which are biodegradation substrates, can be annexed and decomposed by the lysosomal pathway to maintain balanced intracellular lipid metabolism. Decreased intrahepatic autophagic activity, both in vitro and in vivo, can induce excessive lipid deposition [20].
Evidence indicates that glucagon like peptide-1 (GLP-1) protects the liver from cell apoptosis induced by fatty acids though promoting autophagy and suppressing dysfunctional endoplasmic reticulum stress [21,22]. AMP-activated protein kinase (AMPK) is a cellular energy sensor that plays a key role in metabolic disorders and cancer [23]. The AMPK pathway is thought to critically regulate autophagy [24]. More specifically, AMPK activation can inhibit metabolic pathways and activate catabolic pathways to promote effective energy expenditure, which consequently improves how cells adapt to metabolic stress, resulting in increased cell survival [23,25].
Previous studies showed that GLP-1 improves hepatocyte steatosis by inducing autophagy through activating AMPK [26] in mice, suggesting that AMPK protects pancreatic β-cells from high glucose [27]. This evidence provides a new direction for targeting GLP-1 to prevent further deterioration of hepatic steatosis in NAFLD patients. However, the effect of GLP-1 analogs on high-fat diet (HFD)-induced NAFLD in rats has not been investigated. Further, the mechanism underlying liraglutide-induced autophagy is currently unknown. In the current study, we confirmed that liraglutide enhances autophagy in liver tissue and improves hepatic steatosis in a HFD-induced rat model of NAFLD. In addition, we provide evidence supporting the potential mechanism involving activation of the AMPK-mTOR pathway in liver homeostasis.
2Materials and methods2.1AnimalsMale Sprague–Dawley rats (n=50, 5 weeks of age, approximately 100g) obtained from the Laboratory Animal Center of China Medical University were used to establish the NAFLD model. All rats were acclimated for one week prior to experimentation according to a previous protocol published [28]. Briefly, two initial groups of rats were randomly separated. The normal control (NC) group (n=13) was fed chow containing 62% carbohydrates, 10% fat, and 28% protein; the HFD group (n=37) was fed chow containing 34% carbohydrates, 52% fat, and 14% protein. Five rats from each group were randomly selected to confirm successful establishment of NAFLD after 12 weeks of feeding. (All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health.)
Rats in the HFD group were randomly subdivided into four groups: HFD, 50L, 100L, and 200L (n=8 in each group). Rats in the HFD group were continually fed a HFD and were given 0.5ml/kg of normal saline as needed. Rats in the 50L, 100L, and 200L groups continued to receive HFD with different doses of liraglutide (L) (50, 100, or 200μg/kg) (Victoza, Novo-Nordisk A/S, Denmark), respectively. The rats in the NC group (n=8) continued to receive normal chow and were injected with 0.5ml/kg of normal saline. Saline and liraglutide were injected subcutaneously at 8:00am and 8:00pm every day for 4 weeks.
2.2Cell culture and treatmentHepG2 cells were cultured at 37°C in a humidified chamber with 5% CO2 and maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco) containing 10% heat-inactivated fetal bovine serum (FBS, Gibco). There were a total of six cell culture groups: Control group treated with serum-free bovine serum albumin (BSA; Sigma–Aldrich, USA) medium; PA group treated with or without 400M palmitate fatty acid (PA) for 24h; and experimental groups treated with 400M PA and either 10, 50, l00, or 500nmol/L liraglutide for 24h, respectively. These cells were divided into four groups: normal HepG2 cells (BSA), treated with PA (PA), treated with PA and liraglutide (100G), and Compound C for 30min and then treated with PA and liraglutide (100G+C). Command C is an AMPK pathway inhibitor.
2.3Western blot analysisLiver specimens and whole-cell extracts were homogenized and centrifuged in RIPA buffer (Beyotime Institute of Biotechnology, China) containing a protease inhibitor cocktail with 1mmol/L phenylmethanesulfonyl fluoride (Beyotime) on ice. Protein lysates were quantified using the BCA assay as previous described [28] (Pierce, USA). 50μg of protein for each sample were separated by 8–10% SDS-PAGE and transferred to PVDF membranes (Millipore, USA). The membranes were blocked with 5% BSA and then incubated with primary antibodies against microtubule-associated protein LC3, Beclin1, AMPK, phosphorylated (p)-AMPK, TSC1, mTOR, and p-mTOR (all of which were used at 1:1000 and purchased from Cell Signaling Technology, USA) and GAPDH (used at 1:1000; Santa Cruz, CA, USA) at 4°C overnight. The horseradish peroxidase-conjugated anti-rabbit, anti-goat, or anti-mouse secondary antibodies (1:5000, all from Santa Cruz Biotechnology, Inc.) were added at room temperature for 2h after 3 washes (10min each). The immunological complexes were visualized with Micro Chemi 4.2 (DNR Bio-Imaging Systems Ltd., Jerusalem, Israel). Quantity One Software (Bio-Rad, USA) was used to quantify the protein band intensity.
2.4Real-time RT-PCRTotal RNA from the frozen liver specimens (100mg) and cell cultures were isolated using Trizol reagent (Takara Biotechnology Co., Ltd.). The PrimeScriptTM RT reagent kit was used to synthesize cDNA (Takara Biotechnology Co., Ltd., Dalian, China).
Real-time RT-PCR analysis was performed with a Thermal Cycler Dice Real Time detection System (Takara Bio Inc., Japan) using the SYBR Premix Ex Taq II kit (Tli RNaseH Plus) (Takara Biotechnology Co., Ltd.). Gene-specific primers for GFAP and GAPDH were purchased from Takara Biotechnology Co., Ltd. The following PCR protocol was used for all genes: reverse transcription step for 15min at 37°C, then denaturation at 85°C for 5s, 4°C for 7min, followed by an additional 40 cycles of amplification and quantification (5s at 95°C; 30s at 60°C; 30s at 60°C). mRNA expression was normalized to GAPDH as a housekeeping gene. The primers were designed (Primer Premier 5.0) and synthesized (Takara Biotechnology Co., Ltd. Dalian, China). For each gene, the relative change of mRNA in the samples was calculated by subtracting GAPDH Ct values from Ct values for the gene of interest using the 2−ΔΔCt method.
2.5Electron microscopyFor transmission electron microscopy (TEM), liver specimens were fixed with 2.5% glutaraldehyde in a 0.1M sodium cacodylate, pH 7.4, buffer at 4°C and then minced into small (1mm3) fragments. After washing with 0.1M phosphate buffered saline (PBS) three times for 15min each, the liver tissue samples were fixed in 1% osmium tetroxide (OSO4) for 1h, followed by washes in 0.1M PBS. Liver tissues were gradually dehydrated in ethanol solutions of 20%, 50%, 70% and 90% and embedded in Epon 812 epoxy resin before ultrathin sectioning. The ultrastructure of the sections was visualized using a JEM-1200EX electron microscope (JEOL Co., Japan) in the TEM laboratory of China Medical University.
2.6Hematoxylin and eosin (HE) StainingThe liver tissues were fixed at 5mm from the edge of the right lobe of the liver and immersed in 4% paraformaldehyde. Then the tissues were dehydrated, waxed, embedded, and sliced for HE staining. Samples were dewaxed, benzenes were removed, hydrated, hematoxylin stained, washed, eosin stained, dehydrated, and imaged. According to the 2010 guidelines for the diagnosis and treatment of NLFD liver histopathological sections were scored based on the NAS system (0 to 8 points): (1) hepatocellular steatosis: 0 points (<5%); 1 point (5–33%); 2 points (34–66%); 3 points (>66%); (2) Inflammation within the lobules (inflammatory necrosis at 20-fold microscopy): 0 points (none), 1 point (<2), 2 points (2–4), 3 points (>4); (3) liver cell ballooning: 0 points, no; 1 point, rare; 2 points, more common.
2.7Statistical analysisAll data are expressed as the mean±standard deviation (SD) and calculated with one-way analysis of variance (ANOVA) followed by a Newman–Keuls post-hoc test (SPSS 17.0). Results of comparisons were considered significantly different if the p value was<0.05.
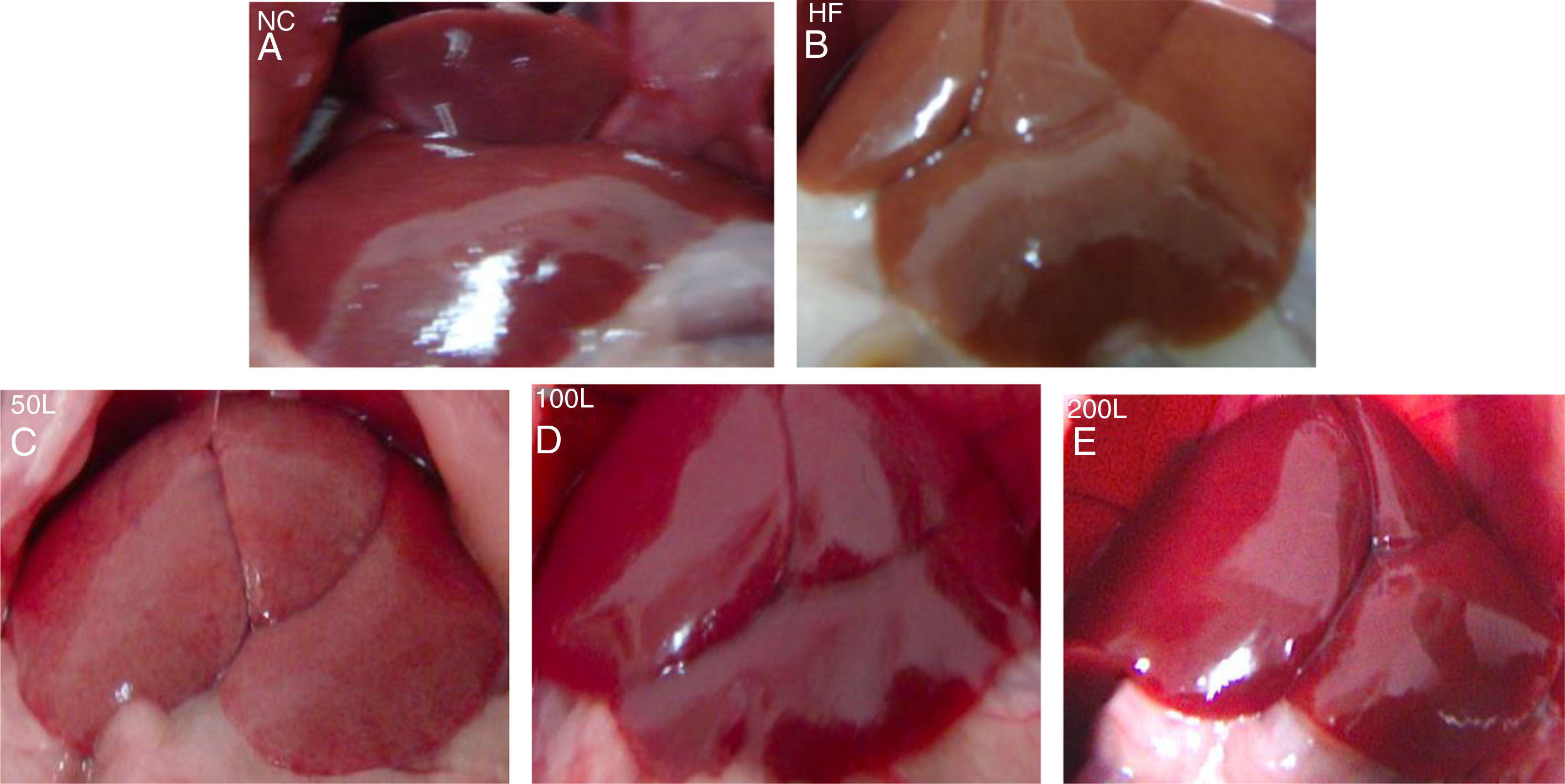
3Results3.1Liraglutide improves hepatic histology during NAFLD developmentAfter 16 weeks of HFD or normal chow diet we examined the histological features of the livers from NAFLD and NC mice. The liver surfaces were greyish yellow and the edges were blunt and thick in the HFD group compared to the NC group, in which the livers were bright red with sharp edges. Liraglutide dose-dependently ameliorated the pathological hepatic histology in the HFD group (Fig. 1A–E).
, D. 100L group: middle dose liraglutide intervention group (100μg/kg), E. 200L group: high-dose liraglutide intervention group (200μg/kg).")
The general changes in liver tissue after 16 weeks of HFD. A. NC group, B. HFD control group, C. 50L group: low dose liraglutide intervention group (50μg/kg), D. 100L group: middle dose liraglutide intervention group (100μg/kg), E. 200L group: high-dose liraglutide intervention group (200μg/kg).
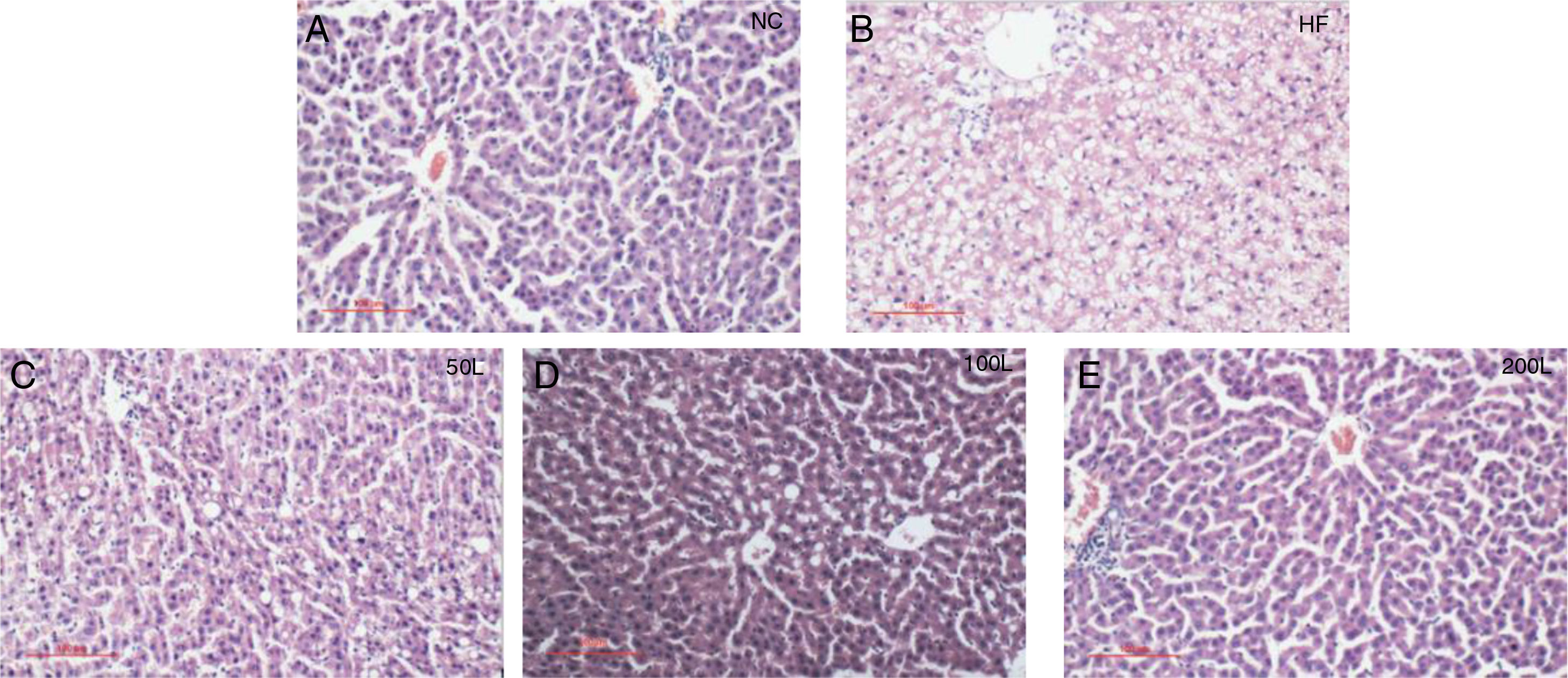
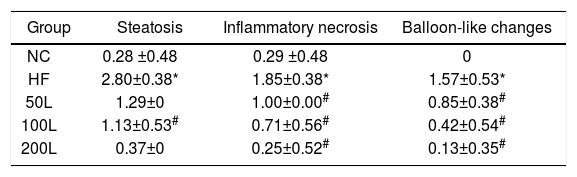
As shown in Fig. 2A–E, liraglutide improved the hepatic histology by significantly reducing both fatty droplets and inflammatory foci number. NAS quantification of liver sections further confirmed the beneficial effects of liraglutide treatment on hepatic histology (Table 1). Compared to the NC rats, the HFD rats had more hepatocellular steatosis (P<0.05), but liraglutide dose-dependently and significantly decreased steatosis, inflammation, and ballooning in the HFD group compared to the NC group (P<0.05).
; B. Rat liver histopathological NAS scores; C. 50L group: low dose liraglutide intervention group (50μg/kg), D. 100L group: middle dose liraglutide intervention group (100μg/kg), E. 200L group: high-dose liraglutide intervention group (200μg/kg).")
Histopathological changes in rat livers 16 weeks after HFD. A. Histopathological changes (× 200); B. Rat liver histopathological NAS scores; C. 50L group: low dose liraglutide intervention group (50μg/kg), D. 100L group: middle dose liraglutide intervention group (100μg/kg), E. 200L group: high-dose liraglutide intervention group (200μg/kg).
The NAS scores of hepatic pathological sections after 16 weeks of HFD
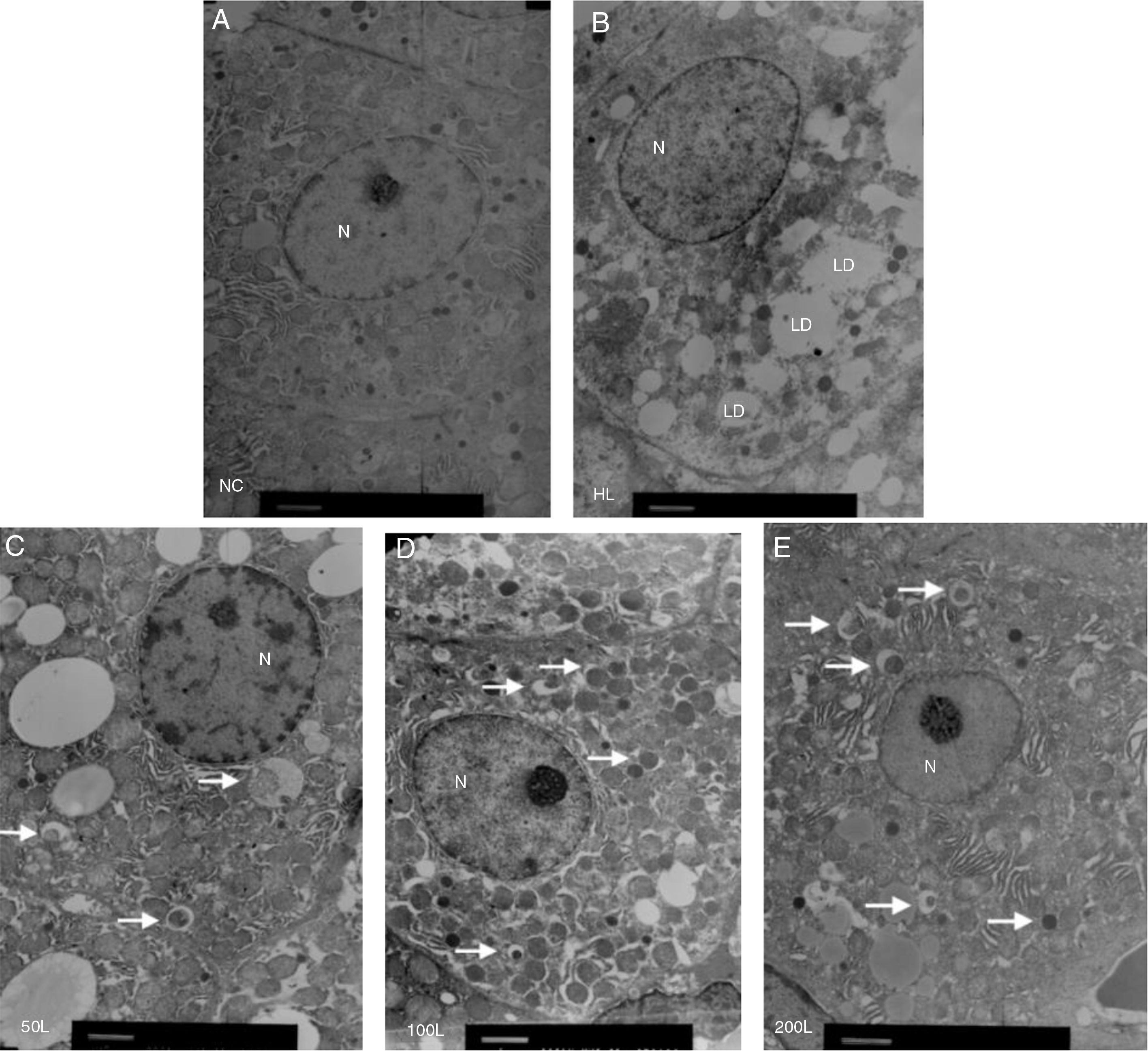
We used electron microscopy to measure autophagosomes in each group. We found that the number of autophagosomes in the HFD group was significantly lower compared to the NC group. Liraglutide treatment dose-dependently increased the number of autophagocytic bodies in the HFD group compared to the NC group (Fig. 3A and B).
, D. 100L group: middle dose liraglutide intervention group (100μg/kg), E. 200L group: high-dose liraglutide intervention group (200μg/kg). N, nucleus; LD, lipid droplet; →, autophagy body.")
Ultrastructure of hepatocytes observed by electron microscopy. A. NC group, B. HF group, C. 50L group: low dose liraglutide intervention group (50μg/kg), D. 100L group: middle dose liraglutide intervention group (100μg/kg), E. 200L group: high-dose liraglutide intervention group (200μg/kg). N, nucleus; LD, lipid droplet; →, autophagy body.
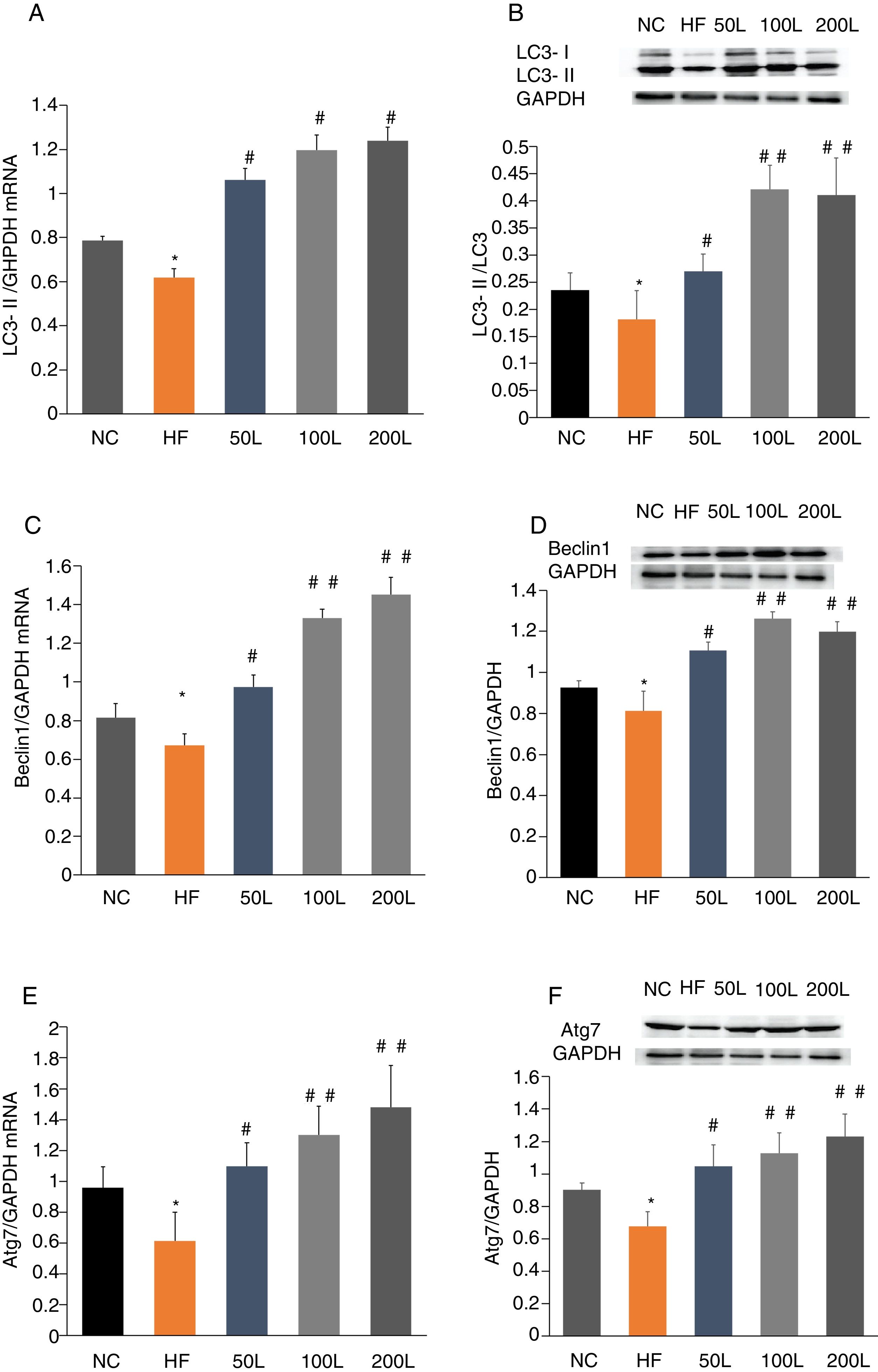
To further determine whether liraglutide improves autophagy in HFD-induced NAFLD rats, we measured the expression of autophagy-related proteins, LC3, Beclin1 and Atg7, in liver tissues. We found that LC3, Beclin1 and Atg7 mRNA levels and protein expression were significantly reduced in the HFD group and that liraglutide treatment dramatically increased expression of these autophagy markers (Fig. 4A–F).

Liraglutide reversed the decrease in autophagy related proteins in HFD-induce NAFLD rats. A and B. LC3II mRNA expression and LC3II/LC3I protein were assessed by real-time RT-PCR and Western blot, respectively. C and D. Beclin1 mRNA and protein expression were detected by real-time RT-PCR and Western blot, respectively. E and F. Atg7 mRNA and protein expression were measured by real-time RT-PCR and Western blot, respectively.
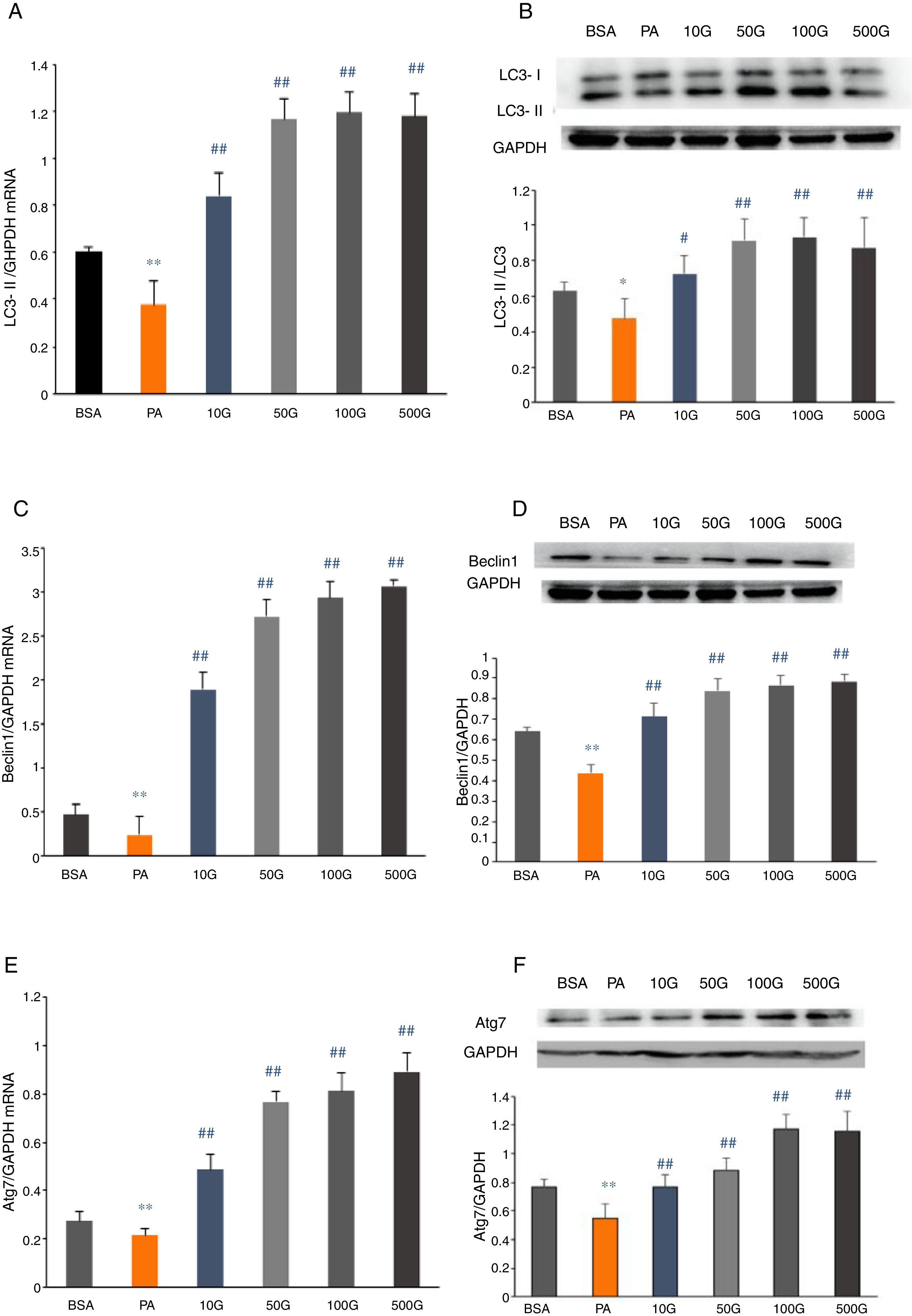
We measured mRNA and protein expression of the autophagy-related proteins, LC3, Beclin1 and Atg7, in HepG2 cells following PA treatment. In the PA group, mRNA and protein expression of LC3, Beclin1 and Atg7 were significantly lower compared to the BSA group (P<0.01). However, liraglutide dose-dependently increased the mRNA and protein levels of LC3, Beclin1 and Atg7 relative to the PA group (P<0.01) (Fig. 5A–F).

Liraglutide reversed the decrease in autophagy related proteins in HepG2 cells. A and B. LC3II mRNA and protein expression were assessed by real-time RT-PCR and Western blot, respectively. C and D. Beclin1 mRNA and protein expression were detected by real-time RT-PCR and Western blot, respectively. E and F. Atg7 mRNA and protein expression were measured by real-time RT-PCR and Western blot, respectively.
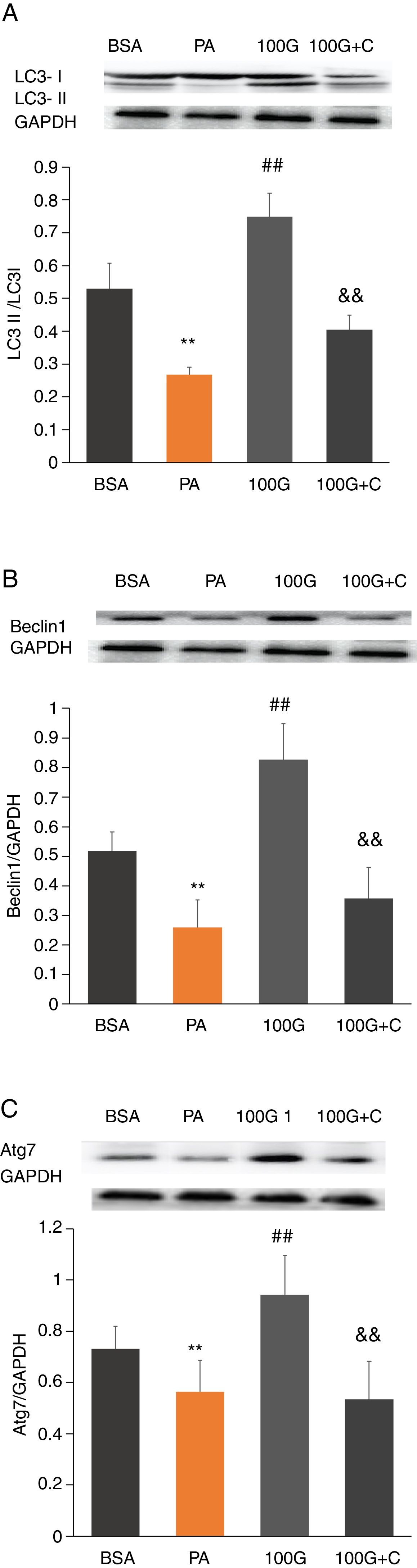
To investigate whether the AMPK pathway was involved in liraglutide-mediated improvement of autophagy after PA treatment, we measured protein levels of LC3, Beclin1 and Atg7 in HepG2 cells. The results showed that the levels of LC3, Beclin1 and Atg7 were significantly higher in the liraglutide intervention group (100G group) compared to the PA group (P<0.01). However, LC3, Beclin1 and Atg7 levels were significantly lower in the inhibition group (100G+C) containing Compound C, PA (400mmol/L) and liraglutide (100nmol/L), compared to the 100G group (P<0.01) (Fig. 6A–C).

The effect of liraglutide on autophagy in HepG2 cells in the presence of the AMPK pathway inhibitor. A. Expression of LC3II/LC3I detected by Western blot with or without the AMPK pathway inhibitor. B. Expression of Beclin1 detected by Western blot with or without the AMPK pathway inhibitor. C. Expression of Atg7 detected by Western blot with or without the AMPK pathway inhibitor.
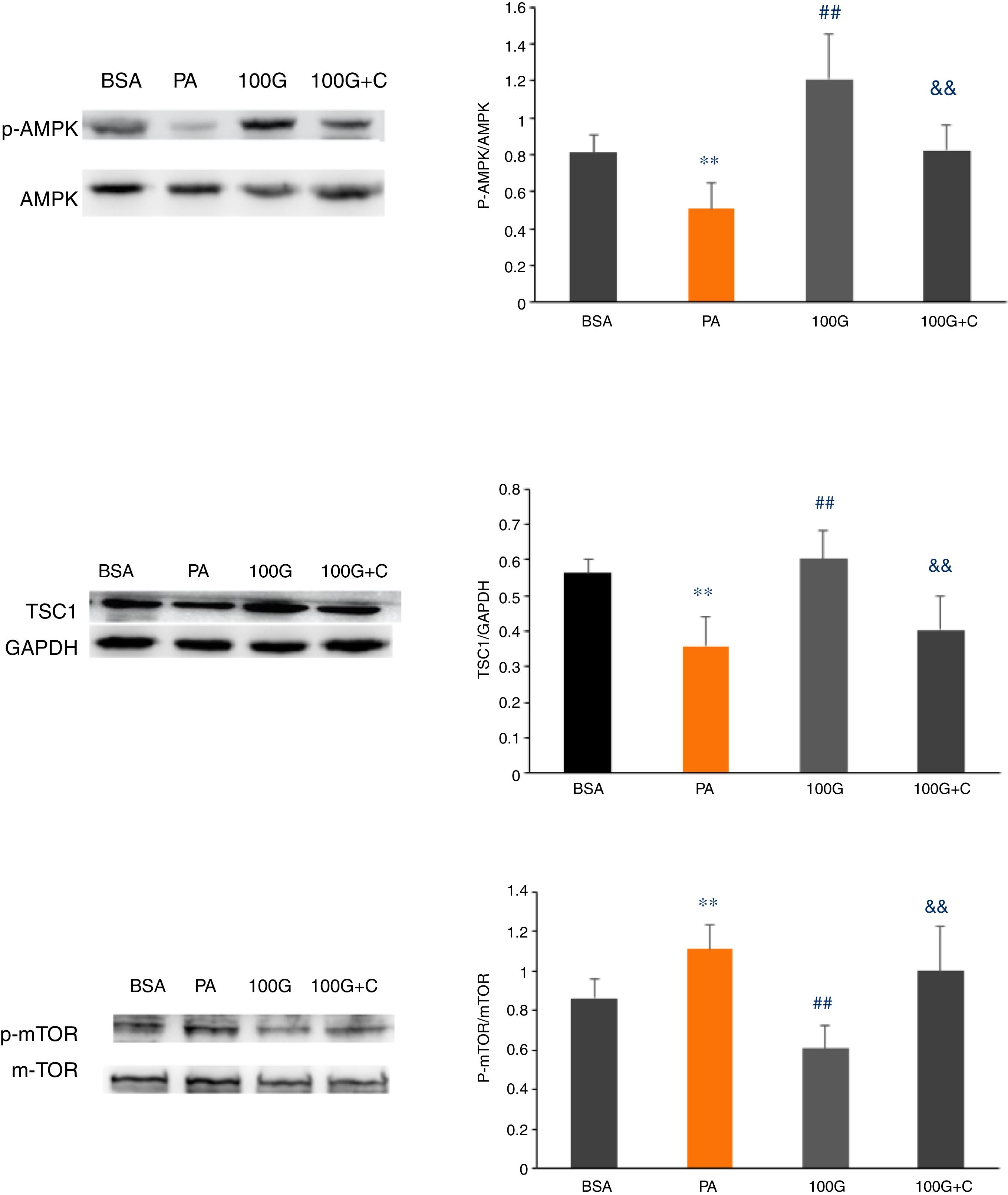
We measured expression of AMPK pathway-associated proteins to confirm that liraglutide-mediated autophagy induced by PA could be reversed by inhibiting the AMPK pathway. As shown in Fig. 6, the levels of p-AMPK/AMPK, TSC1, p-mTOR/mTOR were decreased (P<0.01) in the PA group (400μmol/L PA) compared to the control group (BSA group). Furthermore, the levels of p-AMPK/AMPK, TSC1, and p-mTOR/mTOR were increased in the liraglutide intervention group (100G group) compared to the PA group (P<0.05). The levels of p-AMPK/AMPK, TSC1, p-mTOR/mTOR were significantly lower than those in the 100G group compared to the inhibition group (100 G+C) containing Compound C, PA (400μmol/L), and liraglutide (100nmol/L) (P<0.01) (Fig. 7A–C).
4Discussion
The prevalence of NAFLD has increased worldwide, affecting both adults and children. Liraglutide, a GLP-1 analog, has been reported to decrease lipid deposition and inflammation in hepatocytes [29,30]. However, few studies have investigated whether liraglutide can improve hepatic lipid accumulation in NAFLD. In the current study, we used an established rat model of HFD-induced NAFLD and demonstrated that liraglutide significantly decreased the number of autophagosomes. Liraglutide treatment dose-dependently improved autophagy, which was confirmed by measuring expression of autophagy-related proteins invivo and in vitro. More importantly, the AMPK pathway inhibitor, Compound C, reversed liraglutide's effects on autophagy following PA treatment in vitro, which suggests that liraglutide-induced autophagy is mediated by the AMPK signaling pathway.
Autophagy is an intracellular pathway that maintains normal cellular function by promoting turnover of long-lived proteins [31]. Studies have demonstrated that enhanced autophagy can rescue pancreatic β-cells from glucotoxicity, and inhibition of autophagy augments caspase-3 activation [32], suggesting that autophagy might protect against Type 2 Diabetes. Previous evidence indicated that autophagy is suppressed in the presence of hyper-insulinemia induced by HFD in mice [33], which was consistent with our results; however, we note that we used a different animal model in our study. Our results indicate that autophagy was significantly decreased in the NAFLD rat model, since the autophagy-related proteins, LC3, Beclin1 and Atg7, were significantly reduced at both the mRNA and protein levels. We also noticed that autophagy-related protein levels were decreased in palmitate-induced lipotoxicity in HepG2 cells in vitro, which mimics the pathogenic features of the NAFLD model in vivo.
Liraglutide, a GLP-1 analog, regulates β-cell mass via multiple pathways [34–36]. Liraglutide was proven to decrease lipid accumulation in the steatotic LO2 cell model [37] and protect pancreatic β-cells from high glucose by enhancing autophagy via AMPK [26]. It is also known that GLP-1 exerts protective effects on hepatic steatosis [38]. In the present study, we found that the general appearance, histopathological changes, and the number of autophagic bodies in the livers of NAFLD rats improved after liraglutide treatment. Further evidence showed that liraglutide does-dependently increased the expression of autophagy-related proteins in rats fed a HFD and in HepG2 cells treated with PA. These results suggest that GLP-1 ameliorates HFD induced NAFLD by activating autophagy.
Existing research shows that the AMPK/mTOR pathway regulates downstream signaling to trigger autophagy [39]. AMPK could negatively affect liraglutide-induced increases in cell viability and autophagy to protect insulin-1 pancreatic β-cells from glucotoxicity in rats [27]. In addition, AMPK/mTOR signaling was involved in hepatic lipid metabolism induced by GLP-1 [26].
Our data revealed that Command C, an AMPK pathway inhibitor, reversed the enhanced autophagy induced by liraglutide in HepG2 cells treated with PA, which suggests that liraglutide intervention activates AMPK and up-regulates autophagy. Thus, we conclude that the AMPK pathway plays an important role in regulating autophagy induced by liraglutide.
In conclusion, the current study demonstrates that liraglutide can improve hepatic steatosis via activating the AMPK pathway. These data suggest that GLP-1 may play a protective role in several models of NAFLD and that modulation of AMPK could be a potential target for lipid metabolic disorders.AbbreviationsAMPK adenosine 5′-monophosphate (AMP)-activated protein kinase autophagy related gene 7 heterozygous disruption of beclin1 cyclic adenosine monophosphate glucagon-like peptide-1 micro-tubule-associated protein1 light chain 3 the mammalian target of rapamycin nonalcoholic fatty liver disease palmitate type 2 diabetes mellitus tuberous sclerosis-1
Description of author roles in manuscript creation: Jian Du designed the experiment. Yini He, Na Ao and Jing Yang performed the performed experiments, Xiaochen Wang and Shi Jin processed the data, Yini He wrote the paper, and Jian Du modified the paper.
FundingThe Science and Technology Agency of Liao Ning (20170520272); The Hall Education of Liaoning (L2015567) (LQNK201715).
Conflict of interestAll authors declare no conflicts of interest.
We are grateful for the reagents and technical support provided by the Center Laboratory of The First Hospital and The Laboratory Animal Center of China Medical University.



