










Acute liver injury (ALI) is characterized by massive hepatocyte death with high mortality and poor prognosis. Hepatocyte pyroptosis plays a key role in the physiopathological processes of ALI, which can damage mitochondria and release NLRP3 inflammasome particles, causing systemic inflammatory responses. Z-DNA Binding Protein 1 (ZBP1) is a sensor that induces cell death. Here, we investigated whether ZBP1 participates in hepatocyte pyroptosis and explored the possible pathogenesis of ALI.
Materials and MethodsHepatocyte pyrotosis was induced with lipopolysaccharide (LPS) and nigericin (Nig), and the expression of Zbp1 (ZBP1) was examined by western blot analysis and RT-qPCR. Further, we transfected AML-12 (LO2 and HepG2) cell lines with Zbp1 (ZBP1) siRNA. After ZBP1 was silenced, LDH release and flow cytometry were used to measure the cell death; Western blot analysis and RT-qPCR were used to detect the marker of NLRP3 inflammasome activation and pyroptosis. We also detected the expression of mitochondrial linear rupture marker phosphoglycerate mutase family member 5 (PGAM5) using western blot analysis and reactive oxygen species (ROS) using the DCFH-DA method.
ResultsThe expression of ZBP1 was up-regulated in LPS/Nig-induced hepatocytes. Si-Zbp1 (Si-ZBP1) inhibited NLRP3 inflammasome activation and pyroptosis in LPS/Nig-induced hepatocytes. Moreover, ZBP1 silencing inhibited the expression of PGAM5 by reducing ROS production.
ConclusionsZBP1 promotes hepatocellular pyroptosis by modulating mitochondrial damage, which facilitates the extracellular release of ROS.
Acute liver injury (ALI) is a rapidly developing, complexly induced, and clinically significant liver dysfunction disease [1]. On the one hand, its pathogenesis is due to direct damage to hepatocytes by viruses, drugs, etc. On the other hand, immune-inflammatory injury is mediated by endotoxins. The pro-inflammatory cascade occurs early and is important throughout the course of the disease, ultimately causing systemic multi-organ damage or failure [2]. Recent studies have found that hepatocyte pyroptosis releases NLRP3 inflammasome particles, inducing cytokine storms and systemic inflammatory responses and accelerating the progression of ALI [3]. Hepatocyte pyroptosis [4] is a pro-inflammatory programmed cell death (PCD) form that is distinct from apoptosis and necroptosis. It depends on the activation of caspase-1 mediated by inflammasomes, promoting the release of transmembrane gasdermin D (GSDMD) N-terminal domain protein, which results in membrane pore formation. This process is characterized by cell swelling, pore formation, DNA fragmentation, and the release of cellular contents [5]. Inflammasomes are cytosolic multi-protein complexes that typically recruit adaptor proteins such as apoptosis associated speck-like protein containing a CARD (ASC) via inflammasome sensor molecules like NLR family pyrin domain containing 3 (NLRP3). This recruitment leads to the activation and cleavage of pro-caspase-1, which promote the massive release of mature interleukin 1β (IL-1β) and IL-18. While NLRP3 inflammasome help eliminate infected cells and tumor cells, their abnormal or excessive expression can cause harmful, intense inflammatory responses in the host [6]. Therefore, a deeper understanding of the pathogenic mechanisms of hepatocyte pyroptosis is crucial for preventing and effectively treating ALI.
Mitochondria play a crucial role in regulating NLRP3 inflammasome activation [7]. Reactive oxygen species (ROS) from damaged mitochondria serve as the main activators of NLRP3 inflammasome during acetaminophen (APAP)-induced liver injury, and mitochondria act as docking sites for inflammasome assembly [8]. PGAM family member 5, mitochondrial serine/threonine protein phosphatase (PGAM5) localizes to the outer-inner mitochondrial membrane contact sites, and its activation leads to linear rupture of the mitochondrial cristae [9]. Downregulation of PGAM5 reduces the NLRP3/caspase-1/IL-1β pathway induced by traumatic brain injury, alleviating neuroinflammation and post-injury neuronal damage [9]. However, the regulatory networks of mitochondrial damage need to be further elucidated.
Z-DNA binding protein 1 (ZBP1), also known as DAI [10] or DLM-1 [11], was initially recognized as a key innate immune sensor for viral and endogenous nucleic acid ligands [12]. Recent studies show that LPS induces pulmonary inflammation and injury by activating ZBP1-mediated programmed necroptosis and promoting the secretion of pro-inflammatory cytokines from macrophages [13]. During nonylphenol-induced liver injury, lncRNA PVT1 promoted liver damage by increasing ZBP1 promoter methylation [14]. Therefore, ZBP1 is considered an important regulator of PCD. Additionally, ZBP1 can suppress NLRP3-mediated pancreatic acinar pyroptosis and inflammation in acute pancreatitis by downregulating tRF3-Thr-AGT [15]. Meanwhile, it has been reported that ZBP1 regulates the release of mitochondrial DNA (mtDNA) from tumor cells following radiation therapy, thereby mediating tumor cell death [16]. Thus, lipopolysaccharide (LPS) plus nigericin (Nig) [17] were used to induce NLRP3 inflammasome activation-associated hepatocyte pyroptosis to study whether ZBP1 played a crucial role as a key target protein in ALI.
In the present study, we investigated a novel role of ZBP1 in NLRP3 inflammasome activation-induced cell death and provided a potential therapeutic target for ALI caused by NLRP3 inflammasome activation-associated hepatocyte pyroptosis.
2Material and Methods2.1Cells cultureMouse AML-12, Human LO2, and human hepatoblastoma HepG2 were purchased from the iCell Bioscience (Shanghai, China). The cells were cultured at a constant-temperature incubator (37°C and 5 % CO2) in RPMI 1640 or DMEM medium (Gibco, USA) supplemented with 10 % fetal bovine serum (Gibco, USA) and 1 % penicillin-streptomycinin (Gibco, USA). In addition, the cultivation of AML-12 requires the addition of ITS (Insulin+Transferin+Selenium).
2.2Cells treatmentStandardizing the activation of NLRP3 inflammasome requires two signal steps, namely preprocessing and activation. Escherichia coli 055:B5 lipopolysaccharide (LPS; Sigma, USA) first promotes the transcriptional expression of NLRP3 and pro-IL-1β/pro-IL-18; Furthermore, porogenic toxins such as nigericin (Nig; Invivogen, USA) promote the assembly of inflammasome and promote the release of IL-1β/IL-18.
AML-12 cells were treated with LPS (1 μg/mL) for 22 h, followed by the addition of Nig (0, 2.5, 5 and 10 μM) to the culture for 2 h of further treatment. In the transfection experiment, the cells were treated with LPS (1 μg/mL) for 22 h, followed by the addition of Nig (5 μM) to the culture for 2 h of further treatment.
LO2 cells were treated with LPS (1 μg/mL) for 22 h, followed by the addition of Nig (0, 10, 20 and 40 μM) to the culture for 2 h of further treatment. In the transfection experiment, the cells were treated with LPS (1 μg/mL) for 22 h, followed by the addition of Nig (20 μM) to the culture for 2 h of further treatment.
HepG2 cells were treated with LPS (1 μg/mL) for 22 h, followed by the addition of Nig (0, 5, 10 and 20 μM) to the culture for 2 h of further treatment. In transfection experiments, the cells were treated with LPS (1 μg/mL) for 22 h, followed by the addition of Nig (5 μM) to the culture for 2 h of further treatment.
2.3CCK-8 viability assaysThe Cell Counting Kit-8 (CCK-8; Meiluncell, China) was used to assess the viability of the three cells. The original medium was aspirated. The CCK-8 mixture (CCK-8 reagent: medium=1:10) was added to the cells and incubated for 2 h. The absorbance was determined at an optical density of 450 nm by an enzyme marker.
2.4Giemsa's stainAfter cells treatment, the 24-well plates were removed. Pre-chilled phosphate buffer solution (PBS; Meiluncell, China) was added to wash the cell surface. Add 0.5 mL of 4 % paraformaldehyde into each well for fixation. Wash three times with PBS. 0.3 mL Giemsa's stain solution (Phgene, China) was placed. Stain for 10 minutes at room temperature (RT) on a shaker. Then, gently rinse the plate under running tap water until the background becomes lighter. Inverted microscope was observed and photographed (400 ×).
2.5RT-qPCRExtraction of total mRNA was conducted using TRIzol reagent (Invitrogen, USA). RNA concentration and purity were tested. RNA was reverse transcribed to cDNA according to the HiScript® III All-in-one RT kit (Vazyme, Wuhan, China). Forty amplification cycles (denaturation, annealing, extension) were performed using an ABI 7500 PCR instrument to apply qRT-PCR analysis. The relative expression level was calculated using the 2−△△Ct method. GAPDH was used as an internal reference. Primers were synthesized by IGE BIOTECHNOL (Guangzhou, China) and the primer sequences are shown (Tables 1A and 1B) .
2.6Western blot analysisThe cells were lysed on ice using RIPA buffer (Beyotime, China) containing PMSF. The supernatant was collected to quantify the total protein concentration using the BCA protein assay kit (Beyotime, China). SDS-PAGE was used to separate the denatured proteins and transfer them to PVDF membranes (Millipore, USA). The membranes were sealed with 5 % skim milk powder for 2 h at RT. Then, incubate the indicated antibodies overnight at 4°C. After washing the strips with PBST, they were incubated with a secondary antibody (1:10,000) of the corresponding species at RT. The resulting blots were visualized under a Maging system (Bio-rad, USA) using enhanced chemiluminescence (ECL; Meiluncell, China). The following antibodies were used for western blotting: ZBP1 (1:1000; ZEN BIO, 856002), NLRP3 (1:1000; Affinity, DF7502), ASC (1:1500; ZEN BIO, 340097), GSDMD (1:1000; Affinity, AF4012), caspase-1 (1:2000; ZEN BIO, 342947), caspase-1 p20 (1:1000; Wanleibio, WL03450) and PGAM5 (1:1500; Affinity, DF13355).
2.7Zbp1 (ZBP1) siRNA transfectionTransfection was performed using the Lipofectamine 3000 Transfection Reagent (Thermo, USA). Cells are inoculated in 12-well plates. Each well of cells needs to add 60 pmol siRNA as the transfection amount. Stable incubation for 12 h. The siRNA primer sequences are shown (Tables 2A and 2B).
Mouse-derived siRNA primer sequences from Guangzhou Ige BIOTECHNOLOGY Co., Ltd.
The experiment was first divided into control group, si-nc group, si-Zbp1-1 group, si-Zbp1-2 group and si-Zbp1-3 group to explore the best sequence of ZBP1 knockdown. Then further divided into si-nc group, si-nc+LPS/Nig group and si-Zbp1+LPS/Nig group.
Human-derived siRNA primer sequences from Guangzhou RiboBio Co., Ltd.
| Name | Primer sequences (5ʹ–3ʹ) |
|---|---|
| si-ZBP1-1 | GGAACATCATTACAAGACA |
| si-ZBP1-2 | CGAGACTTGTACAGGATGA |
| si-ZBP1-3 | GGATGAGCAGTCCAAAGCA |
The experiment was first divided into a control group, si-NC group, si-ZBP1-1 group, si-ZBP1-2 group and si-ZBP1-3 group to explore the best sequence of ZBP1 knockdown. Then further divided into si-NC group, si-NC+LPS/Nig group and si-ZBP1+LPS/Nig group.
The three cell lines were stably cultured in 96-well plates for 24 h. Treat with siRNA for 12 h. The groups continued to stimulate the cells with the optimal concentration of LPS/Nig for 24 h. Collect the culture medium of the cells. The LDH cytotoxicity assay kit (Beyotime, China) was used to measure the LDH release. The absorbance was read at 490 nm and 570 nm using an enzyme marker.
2.9Annexin V-FITC/PI+ assayRemove the 12-well plates, aspirate the medium into an EP tube and label it. Wash the cell with PBS and add 0.25 % trypsin (Gibco, USA) for digestion. After digestion was completed, the digestion was terminated with the collected old medium. Cells were collected and centrifuged at 2000 rpm for 10 minutes. Wash the cells again with PBS and remove the supernatant by centrifugation. Add 100 µL cell flow dilution, 2.5 µL Annexin V-FITC and 2.5 µL PI to each tube and mix thoroughly. Take three centrifuge tubes, labeled as blank control tube, Annexin V-FITC alone tube and PI alone tube. Incubate for 10 min at RT protected from light. Then, add 500 μL PBS to each tube and place it on ice. Timely detection by flow cytometry (BD, USA).
2.10DCFH-DA assayThe 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime, China) was diluted 1000-fold with serum-free medium to a final concentration of 10 μmol/L. Wash the cells with PBS. The DCFH-DA solution was added to each well to cover all cells and incubated for 20 min at 37°C in cells incubator. The fluorescence release from different hepatocyte lines in 12-well plates was observed promptly using an inverted fluorescence microscope. The ROS fluorescence intensity in 96-well plates was measured on an enzyme marker with an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
2.11Statistical analysisStatistical analysis was performed using SPSS 23.0 (SPSS, Chicago, USA) and GraphPad Prism 9.0 software (GraphPad Software Inc., La Jolla, CA, USA). Data are presented as mean ± standard deviation (SD). Variables were initially verified by normal distribution. Means of multiple groups were compared using one-way ANOVA, and comparisons between any two groups were further analyzed using Bonferroni t-test. P<0.05 was considered statistically significant.
2.12Ethical statementsThis article is a non-interventional study (cellular experiment). According to the Ethics Committee, ethical approval is not required. Of course, certain ethical principles and norms are strictly followed when conducting cell experiments, and the use of human samples from illegal sources is prohibited.
3Results3.1NLRP3 inflammasome activation-associated hepatocyte pyroptosis is induced by LPS/NigIt was found that NLRP3 inflammasome activation-associated hepatocyte pyroptosis was induced by LPS plus Nig. First, with the gradual increase of Nig concentration, the activity and number of hepatocytes gradually decreased (P<0.01). The cells gradually lost their normal morphology, the cytoplasm became hyaline, and the nucleus staining deepened (Fig. 1A and B). Further, Western blot analysis revealed increased levels of NLRP3 and caspase-1 p20 in LPS/Nig-treated hepatocytes compared to the control and LPS alone group (Fig. 1C). Increased mRNA levels of Il-1β/Il-18 (IL-1β/IL-18) were observed in LPS/Nig-treated hepatocytes compared to the control and LPS alone group (P<0.05) (Fig. 1D). Consistently, we also observed an increase in the protein levels of GSDMD-N (Fig. 1E).
. F Protein level detection of GSDMD-N expression. All experiments were performed three times independently. aP<0.05 vs control group, bP<0.01 vs control group, cP<0.001 vs control group, #P<0.05 vs LPS group.")
NLRP3 inflammasome activation-associated hepatocyte pyroptosis is induced by LPS/Nig. The different hepatocyte lines treated with LPS for 22 h followed by the addition of different concentration of Nig for 2h of further treatment. A Cell Viability detected by CCK-8 assays. B The morphology measured using Giemsa staining. Magnification 400x. C Western blot for the levels of NLRP3, ASC, caspase-1 and caspase-1 p20 expression. D RT-qPCR for relative mRNA level of Il-1β/Il-18 (IL-1β/IL-18). F Protein level detection of GSDMD-N expression. All experiments were performed three times independently. aP<0.05 vs control group, bP<0.01 vs control group, cP<0.001 vs control group, #P<0.05 vs LPS group.
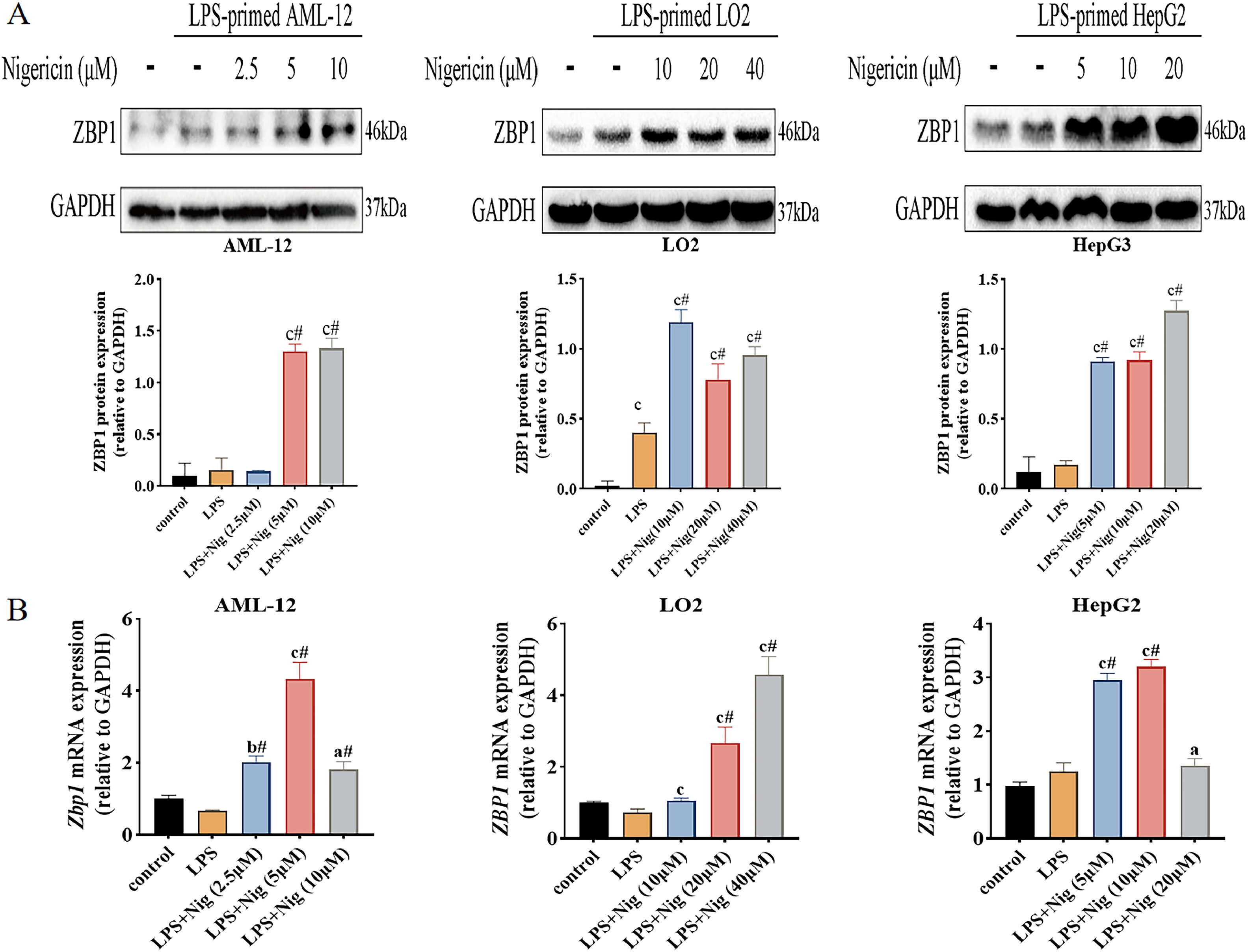
Further, we examined the expression levels of ZBP1 in different hepatocyte lines treated with LPS plus Nig. The results of western blot analysis showed that ZBP1 expression increased in LPS/Nig-treated cells compared to the control and LPS alone group (Fig. 2A); the mRNA levels of Zbp1 (ZBP1) increased in LPS/Nig-treated cells compared to the control and LPS alone group (P<0.05) (Fig. 2B). Summarily, when hepatocyte pyroptosis was successfully induced LPS/Nig, the expression levels of ZBP1 increase.
 level. All experiments were performed three times independently. aP<0.05 vs control group, bP<0.01 vs control group, cP<0.001 vs control group, #P<0.05 vs LPS group.")
ZBP1 expression increases in LPS/Nig-treated different hepatocyte lines. The different hepatocyte lines treated with LPS for 22 h followed by the addition of different concentration of Nig for 2 h of further treatment. A Western blot detection of ZBP1 expression. B RT-qPCR detection of Zbp1 (ZBP1) level. All experiments were performed three times independently. aP<0.05 vs control group, bP<0.01 vs control group, cP<0.001 vs control group, #P<0.05 vs LPS group.
To verify the role of ZBP1 in LPS/Nig-treated different hepatocyte lines. Zbp1 (ZBP1) expression was silenced using siRNAs and silencing efficiency of the three siRNA was evaluated using RT-qPCR. The si-Zbp1-3 (si-ZBP1-3) could effectively silence the mRNA levels of Zbp1 (ZBP1) (P<0.001) (Fig. 3A). Then, the silencing efficiency was further verified by Western blot (Fig. 3B). LDH release assays to assess cytotoxicity showed that combination of LPS and Nig increased LDH release in different hepatocyte lines; silenced-ZBP1 reversed LDH release (P<0.05) (Fig. 3C). Data from flow cytometric analysis revealed that the proportion of Annexin V/PI+ positive cells was more increased in LPS/Nig than in si-nc (si-NC) group (P<0.05); down-regulated ZBP1 blocked the proportion significantly (P<0.05) (Fig. 3D). Thus, ZBP1 silencing could diminishe cytotoxicity and the proportion of PI+ positive cells.
 levels of different hepatocyte lines transfected with siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1). B Further, Western blot for the expression of ZBP1 of cells transfected with or without siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1). C LDH release of LPS/Nig-treated hepatocytes transfected with siRNA (si-nc and si-Zbp1,or si-NC and si-ZBP1). D Flow cytometric analysis for the proportion of Annexin V/PI+ positive cells of LPS/Nig-treated hepatocytes transfected with siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1). All experiments were performed three times independently. *P<0.05 vs si-nc (si-NC) group, ⁎⁎P<0.01 vs si-nc (si-NC) group, ⁎⁎⁎P<0.001 vs si-nc (si-NC) group, #P<0.05 vs si-nc+LPS/Nig (si-NC+LPS/Nig) group.")
ZBP1 accelerates the promotion of LPS/Nig-induced hepatocytes death. A RT-qPCR for Zbp1 (ZBP1) levels of different hepatocyte lines transfected with siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1). B Further, Western blot for the expression of ZBP1 of cells transfected with or without siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1). C LDH release of LPS/Nig-treated hepatocytes transfected with siRNA (si-nc and si-Zbp1,or si-NC and si-ZBP1). D Flow cytometric analysis for the proportion of Annexin V/PI+ positive cells of LPS/Nig-treated hepatocytes transfected with siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1). All experiments were performed three times independently. *P<0.05 vs si-nc (si-NC) group, ⁎⁎P<0.01 vs si-nc (si-NC) group, ⁎⁎⁎P<0.001 vs si-nc (si-NC) group, #P<0.05 vs si-nc+LPS/Nig (si-NC+LPS/Nig) group.
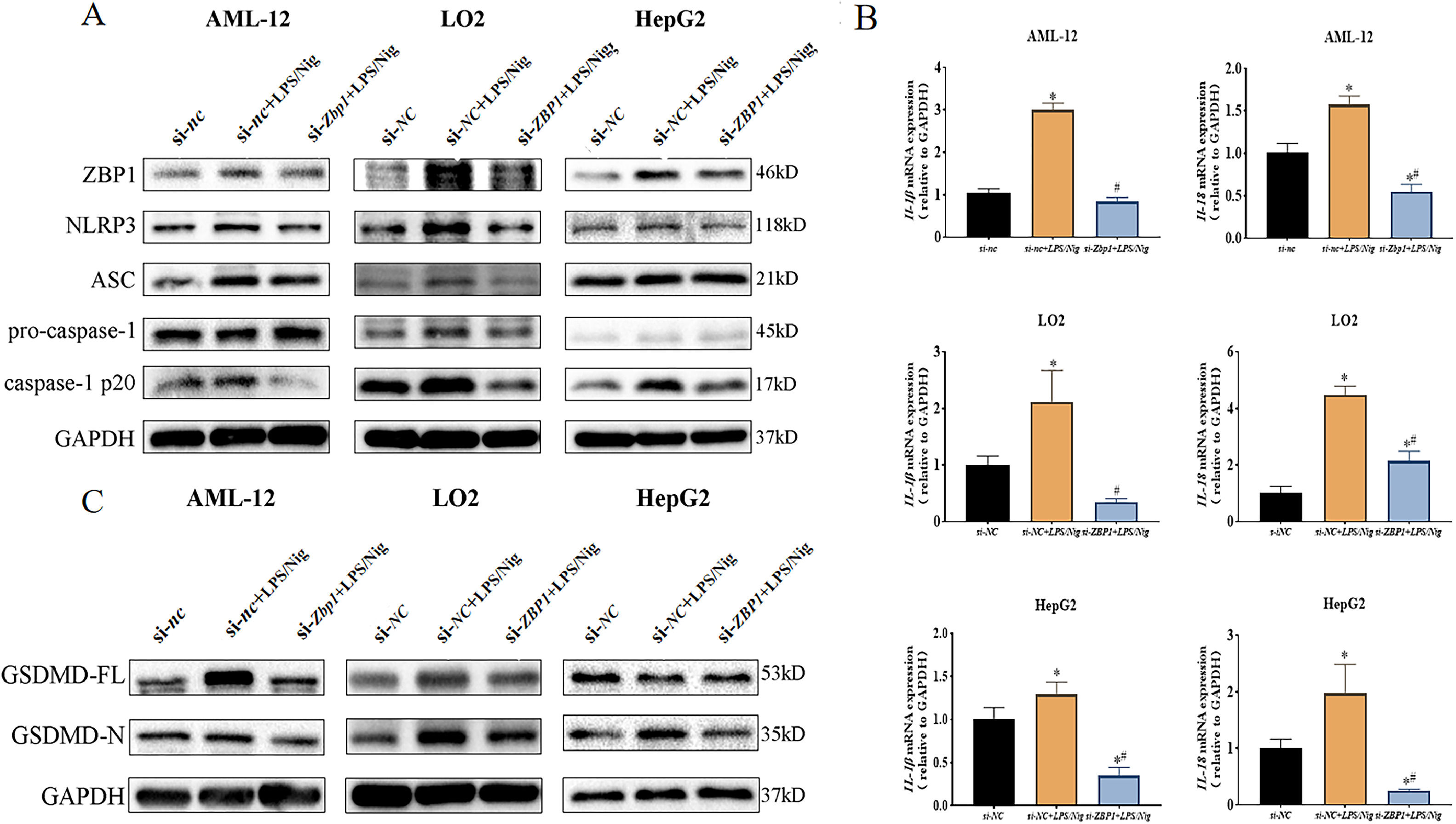
AML-12 cell line was divided into 3 groups (si-nc, si-nc+LPS/Nig and si-Zbp1+LPS/Nig group). LO2 and HepG2 cell lines were divided into 3 groups (si-NC, si-NC+LPS/Nig and si-ZBP1+LPS/Nig group). Data showed that a down-regulation of NLRP3 and caspase-1 p20 expression was induced by si-Zbp1 (si-ZBP1) compared with si-nc+LPS/Nig group (si-NC+LPS/Nig group) (Fig. 4A). Consistently, decreased mRNA levels of Il-1β/Il-18 (IL-1β/IL-18) were observed (P<0.05) (Fig. 4B). In addition, ZBP1 silencing resulted in down-regulated GSDMD-N expression in LPS/Nig-treated hepatocytes (Fig. 4C). Thus, ZBP1 silencing attenuated LPS/Nig-treated hepatocyte pyroptosis via inhibition of NLRP3 inflammasome activation.
 were stably cultured for 24 h after treatment with LPS/Nig. A Western blot for the levels of ZBP1, NLRP3, ASC, caspase-1 and caspase-1 p20. B RT-qPCR for relative mRNA level of Il-1β/Il-18 (IL-1β/IL-18). C Western blot detection of GSDMD-N expression. All experiments were performed three times independently. *P<0.05 vs si-nc (si-NC) group, #P<0.05 vs si-nc+LPS/Nig (si-NC+LPS/Nig) group.")
ZBP1 triggeres hepatocyte pyroptosis through activation of NLRP3 inflammasome. Different hepatocyte lines transfected with siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1) were stably cultured for 24 h after treatment with LPS/Nig. A Western blot for the levels of ZBP1, NLRP3, ASC, caspase-1 and caspase-1 p20. B RT-qPCR for relative mRNA level of Il-1β/Il-18 (IL-1β/IL-18). C Western blot detection of GSDMD-N expression. All experiments were performed three times independently. *P<0.05 vs si-nc (si-NC) group, #P<0.05 vs si-nc+LPS/Nig (si-NC+LPS/Nig) group.
To further explore the functional network, we measured the expression of PGAM5 and intracellular ROS. Western blot analysis showed that PGAM5 was up-regulated in LPS/Nig treated hepatocytes. However, when the expression of ZBP1 was downregulated, the expression levels of PGAM5 decreased (Fig. 5A). Further, DCFH-DA was used to detect intracellular ROS. The fluorescence intensity of DCF was proportional to the expression level of ROS. Immunofluorescence and DCF fluorescence intensity showed that the fluorescence levels of cellular ROS of si-nc+LPS/Nig group (si-NC+LPS/Nig) was higher than that of si-nc (si-NC) group (P<0.01). After ZBP1 was silenced, ROS levels were reduced in AML-12 and LO2 cell lines (P<0.01), while there was a trend of reduced ROS levels in HepG2 cell lines, but no statistical difference (P>0.05) (Fig. 5B and 5C). Thus, ZBP1 silencing inhibited NLRP3 inflammasome activation by mediating the PGAM5/ROS pathway.
 were stably cultured for 24 h after treatment with LPS/Nig. A Western blot detection of PGAM5 expression. B Immunofluorescence staining images (Magnification 100x) and C DCF Fluorescence intensity showing ROS expression. All experiments were performed three times independently. *P<0.05 vs si-nc group (si-NC group), #P<0.05 vs si-nc+LPS/Nig group (si-NC+LPS/Nig group).")
ZBP1 promotes NLRP3 inflammasome activation-associated hepatocyte pyroptosis by regulating PGAM5/ROS pathway. Different hepatocyte lines transfected with siRNAs (si-nc and si-Zbp1,or si-NC and si-ZBP1) were stably cultured for 24 h after treatment with LPS/Nig. A Western blot detection of PGAM5 expression. B Immunofluorescence staining images (Magnification 100x) and C DCF Fluorescence intensity showing ROS expression. All experiments were performed three times independently. *P<0.05 vs si-nc group (si-NC group), #P<0.05 vs si-nc+LPS/Nig group (si-NC+LPS/Nig group).
ALI is one of the most common liver diseases with a complex pathogenesis and characterization, including hepatocyte death, oxidative stress, and inflammation. Numerous studies have shown that hepatocyte pyroptosis is involved in the pathological process of ALI, characterized by rapid plasma membrane rupture and massive release of pro-inflammatory intracellular contents [18]. Inhibiting NLRP3 inflammasome-induced pyroptosis and inflammatory response can prevent LPS/D-GalN-induced ALI [19]. This study found that NLRP3 inflammasome activation and hepatocyte pyroptosis were successfully induced by LPS/Nig3. We were surprised to find that the dose of Nig used to achieve 50 % survival of hepatocytes from different sources was different. This may be related to the species of the cells, the method of immortalization and the culture medium. Interestingly, when cell viability decreased to ∼30 %, the protein expression levels were downregulated. This may be related to the negative feedback regulation of GSDMD [20].
ZBP1 was recently identified as a cytoplasmic sensor for viral infections that regulates cell death, inflammasome activation, and pro-inflammatory responses [21]. ZBP1 is expressed in hepatocytes and regulates hepatitis virus replication in an investigation of woodchuck hepatitis virus modeling viral hepatitis [22]. Moreover, during NP-induced liver injury, lncRNA PVT1 can increase ZBP1 promoter methylation to accelerate liver injury [14]. Further, necrostatin-1 (Nec-1) treatment of ventilator-induced lung injury (VILI) mainly improves lung epithelial cell pyroptosis by inhibiting caspase-1-dependent NLRP3 inflammasome activation and reducing the key pyroptotic mediator GSDMD through the RIPK/ZBP1 pathway [23]. It has been reported that during influenza A virus infection, immune cells abnormally express ZBP1, which is considered a critical upstream regulatory factor of the pyrin domain-containing NLRP3 inflammasome [24]. As previously described, ZBP1 has many biological functions and participates in regulating the progression of various diseases; however, the roles of ZBP1 in ALI are largely unknown.
In recent years, ZBP1 has been used as a potential target in cancer immunotherapy. Activation of ZBP1 ultimately leads to RIPK3-mediated necroptosis in cells [25]. In the current study, we found that ZBP1 could promote hepatocellular pyroptosis through the PGAM5/ROS axis. Our research provides a potential target for ALI treatment. For the first time, we found that ZBP1 expression could be up-regulated in LPS/Nig-induced ALI. ZBP1 silencing could reduce cell death, regulate NLRP3 inflammasome activation-associated hepatocyte pyroptosis, and improve acute liver injury. Mitochondria are the major organelles in which cellular aerobic respiration plays a crucial role in regulating NLRP3 inflammasome activation. PGAM5 is also considered a novel inflammasome and caspase-1 activity regulator, functioning independently of RIPK3. Abnormal activation of PGAM5 can also enhance ROS production and associated oxidative damage. The accumulation of ROS stimulates the persistent opening of mitochondrial membrane pores, leading to reduced mitochondrial membrane potential, respiratory chain abnormalities, mitochondrial protein release, and the induction of cell death. Here, it was found that ZBP1 downregulation could improve mitochondrial damage.
It is necessary to acknowledge the limitations of this work. First, this study may be more oriented to innovative exploration due to the limited amount of literature related to ZBP1 in liver injury; second, the lack of results from animal experiments limits the extension of the molecular mechanism; and lastly, for the current ZBP1 findings, it is still a basic research bias, but its prospect is wide and unlimited. In recent years, it has been found that cell death is no longer a single mode of death but presents a continuous death process. ZBP1 is the hub protein in it. Targeted regulation research will be the key direction.
5ConclusionsTaking together, ZBP1 promotes hepatocellular pyroptosis by modulating mitochondrial damage, which facilitated the extracellular release of ROS. In conclusion, this study extends our understanding of the physiological functions and regulatory pathways of ZBP1 to explore a novel target and mechanism that lead to the inflammatory response and hepatocellular damage for ALI.
Author contributionsYLN and OYS conceived the study and conducted the experiments. XD acquired the data. YSG conducted statistical analyses. CZR and HZL assisted in the experiments. The manuscript was prepared, edited, and reviewed by YSG, YLN and OYS. A final version of the manuscript has been read and approved by all authors.
Data availabilityUpon request, data will be provided.
FundingThis work was supported by the National Natural Science Foundation of China (81803884), Plan on enhancing scientific research in GMU, 2022 Guangzhou Medical University discipline construction projects (02-410-2206013), the 2022 Student Innovation Capacity Enhancement Program Project of Guangzhou Medical University (02-408-2304-19064XM), 2023 City school (college) enterprise joint funding projects (2023A03J0421), the Undergraduate Capacity Enhancement Innovation Project of Guangzhou Medical University (2022JXA003), the Key Medical Discipline of Guangzhou [2021-2023], Doctoral research foundation of North China University of Science and Technolog and the Key Laboratory of Guangdong Higher Education Institutes (2021KSYS009).



