


El síndrome de Brugada es una enfermedad hereditaria caracterizada por una anormalidad electrocardiográfica y un aumento del riesgo de muerte súbita cardiaca. El síndrome de Brugada puede ser causado por la presencia de mutaciones en el gen SCN5A en aproximadamente el 20% de los casos familiares. El gen SCN5A codifica la subunidad α del canal iónico de sodio en las células cardiacas. Estudios realizados durante la última década en genética molecular han permitido identificar 11 nuevos genes con susceptibilidad para síndrome de Brugada además del SCN5A, lo que lleva a pensar que es una enfermedad con heterogeneidad genética y compleja de identificar en la clínica y a nivel molecular en el laboratorio. Una manera de heredar el síndrome de Brugada es por medio de un patrón de transmisión hereditaria autosómica dominante.
Esta breve revisión se enfoca a describir el proceso de diagnóstico de marcadores genéticos en un caso reportado de síndrome de Brugada guiando al lector a través del proceso de identificación de las variantes genéticas responsables del síndrome y a determinar la consecuencia funcional de las mutaciones del canal de sodio sobre la alteración electrocardiográfica.
Brugada syndrome is a genetic disease that is characterized by abnormal electrocardiogram findings and an increased risk of sudden cardiac death. This syndrome is linked to mutations in the SCN5A gene in approximately 20% of Brugada syndrome probands. SCN5A encodes the α subunit of the cardiac sodium channel. Studies conducted over the past decade have identified 11 other Brugada syndrome susceptibility genes besides to SCN5A, pointing to genetic heterogeneity of the syndrome. Transmission of the disease shows an autosomal dominant inheritance pattern.
This brief review focuses on a reported case of sodium channel-mediated Brugada syndrome, guiding the reader through the process of identification of the genetic variants responsible for the clinically-diagnosed syndrome, mutagenesis to clone SCN5A with and without the 2 variants identified and transfection of the 2 variants into TSA201 cells to determine the functional consequence of these genetic variants on sodium channel expression and function.
El síndrome de Brugada (SBr) es una enfermedad hereditaria caracterizada por una anormalidad electrocardiográfica y un aumento del riesgo de muerte súbita cardiaca. El SBr (herencia mendeliana humana, OMIM-601144) presenta varios tipos dependiendo del gen mutado. La primera mutación descrita se identificó en el gen SCN5A (OMIM-600163) ubicado en el brazo corto del cromosoma 3 (3p21). Aproximadamente el20% de los pacientes con SBr presentan mutaciones en este gen. El gen SCN5A codifica para la subunidad α del canal de sodio que determina la fase 0 del potencial de acción de las células cardiacas1. Los estudios realizados en la última década identificaron otros 11 genes responsables del SBr además del SCN5A, señalando la heterogeneidad genética del síndrome. La transmisión genética de la enfermedad presenta un patrón de herencia autosómica dominante2.
Esta revisión se enfoca a describir el proceso de diagnóstico de marcadores genéticos en un caso reportado de SBr guiando al lector a través del proceso de identificación de las variantes genéticas responsables del síndrome y a determinar la consecuencia funcional de las mutaciones del canal de sodio sobre la alteración electrocardiográfica. El proceso de diagnóstico de marcadores genéticos a nivel molecular se realizó utilizado el material genético extraído de células de un paciente con SBr que presentaba un elevado riesgo de sufrir fibrilación ventricular o taquicardia ventricular, pudiendo desencadenar un síncope con muerte súbita3.
Los pacientes que manifiestan clínicamente el SBr presentan generalmente una reducción de la corriente entrante de sodio (INa+) debida a mutaciones en el gen SCN5A. Los fármacos antiarrítmicos utilizados para el tratamiento del SBr tienen un efecto directo sobre los canales de sodio cardiacos.
Para estudiar los mecanismos moleculares e iónicos del SBr ligados a nuevas mutaciones en los canales de sodio cardiacos es necesario al menos estudiar los siguientes 3 procesos: (1) Identificar las mutaciones del gen SCN5A en los pacientes clínicamente diagnosticados con SBr. (2) Determinar los efectos de las mutaciones sobre la INa+, mediante el registro de la corriente macroscópica por el método de fijación de voltaje en célula completa. (3) Determinar el efecto de los fármacos antiarrítmicos utilizados para el tratamiento de arritmias ventriculares sobre la INa+ de los canales de sodio mutados4.
Descripción del caso clínico de los pacientes con síndrome de BrugadaEstudiamos un caso de SBr de tipo hereditario previamente diagnosticado por una institución de cardiología en EE. UU. Todos los familiares relacionados con el paciente se sometieron a un estudio de electrocardiografía (registro de 12 derivaciones) y ecocardiografía para descartar la observación de alguna enfermedad estructural cardiaca. Los criterios para designar el diagnóstico del SBr fueron acordados en función de las anormalidades en el ECG5.
Los pacientes con SBr presentaron al menos 2 de las características clínicas que se mencionan a continuación (alguna de las 2 primeras y cualquiera de las 2 últimas):
- 1
La elevación del segmento ST en las derivaciones precordiales derechas V1 a V3 del ECG.
- 2
La elevación del segmento ST en presencia de fármacos bloqueadores de sodio, «desenmascarando» el SBr.
- 3
Presentar síncopes y arritmias como la fibrilación ventricular, fibrilación auricular, Torsades de pointes (fibrilación ventricular polimórfica) y también la muerte súbita con o sin resucitación. Los pacientes no se incluirán si presentan alteraciones estructurales cardiacas previas.
- 4
Historial clínico, la identificación de familiares del caso índice con el SBr.
La extracción del ácido desoxirribonucleico (ADN) se llevó a cabo a partir de linfocitos sanguíneos por medio del método estándar de extracción de ADN genómico (Gentra System, Puregene). Mediante la reacción en cadena de la polimerasa se amplificaron todos los exones e intrones del gen SCN5A y los productos obtenidos a partir de cada oligonucleótido fueron purificados con un reactivo comercial (ExoSAP-IT, USB). Posteriormente se realizó la secuenciación directa por el método de Sanger en ambas direcciones con el uso del secuenciador de ADN automático ABI PRISM 3100-Avant (Automatic DNA sequencer). Para determinar la prevalencia de las variaciones en la población étnica control se analizaron 400 sujetos anónimos sin problemas cardiovasculares; se presume que el 100% de ellos no presentaron mutaciones en el gen SCN5A y ningún síntoma del SBr.
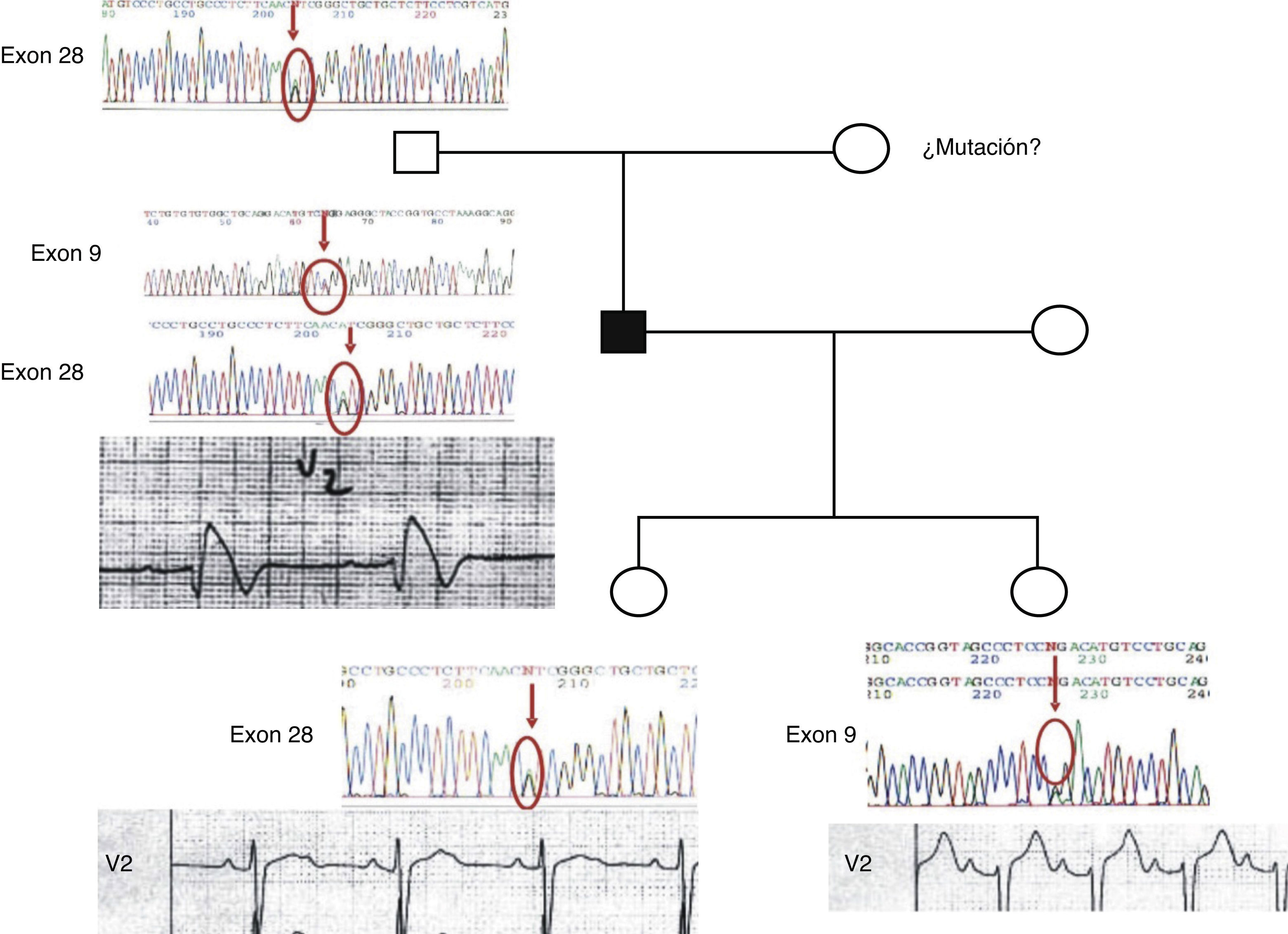
Diagnóstico clínico y molecular genéticoEl caso índice o probando presentó un caso severo de SBr. El análisis del ECG mostró una marcada elevación en el segmento ST, indicador del SBr. Al realizarse el análisis genético molecular se identificaron 2 mutaciones puntuales; una de ellas fue por sustitución de la prolina por la leucina en la posición 336 (P336L) y la segunda mutación fue una sustitución de una isoleucina por una valina en la posición 1660 (I1660V). Dentro del análisis genético del árbol genealógico del probando se identificó la presencia de mutaciones separadas en cada una de las hijas (fig. 1). Las 2 hijas del caso probando muestran cambios menores en el ECG; una de ellas presentó una ligera prolongación del intervalo PR en el ECG con la presencia de la mutación I1660V, y la otra hija muestra una ECG normal con la presencia de la mutación P336L.
Expresión fenotípica diferencial resultado de la presencia de mutación doble en el canal de SCN5A en una familia con síndrome de Brugada.")
Para poder dar una explicación de este fenómeno clínico realizamos un estudio experimental in vitro mediante la expresión de las diferentes mutaciones P336L e I1660V en el gen SCN5A en células TSA201 y registrando la corriente entrante de sodio de manera diferencial.
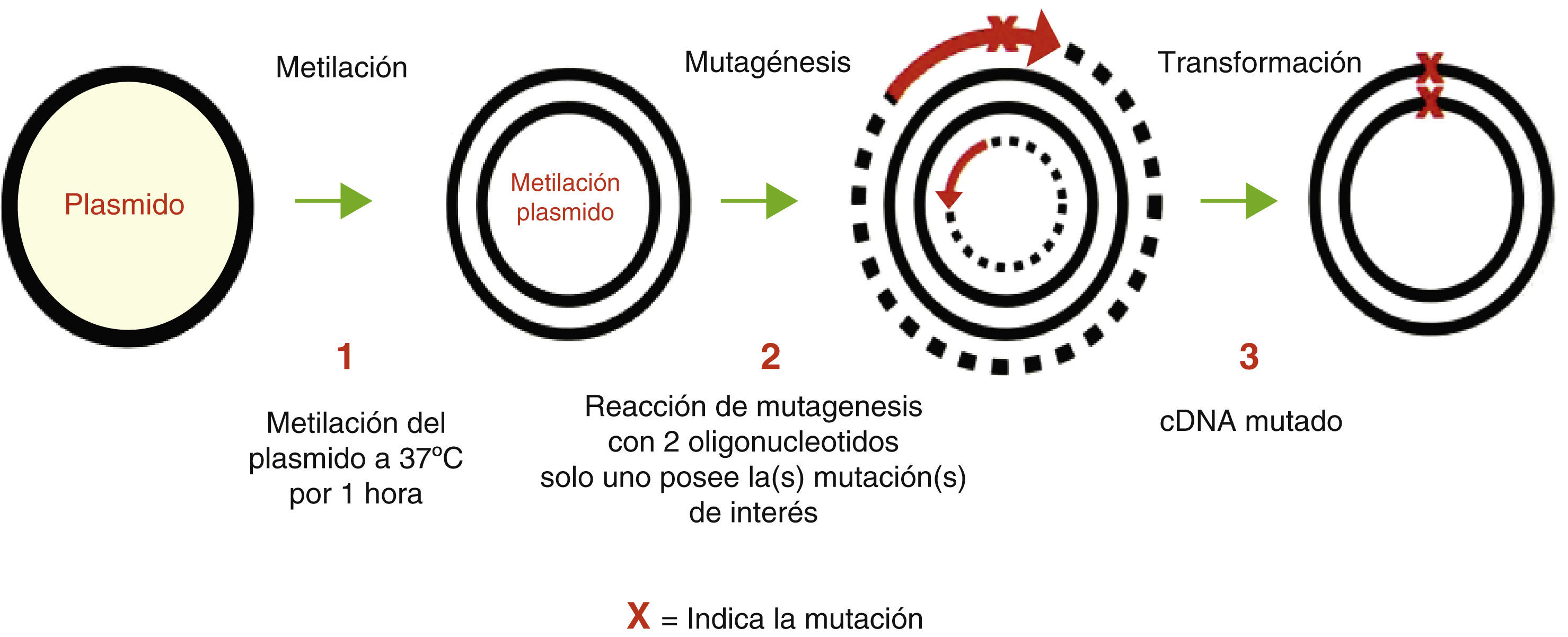
Mutagénesis sitio-dirigida del gen SCN5ALas mutaciones del gen SCN5A encontradas fueron construidas usando el sistema de mutagénesis sitio-dirigida (fig. 2) mediante el sistema Gene TaylorTM (Invitrogen Corp) usando el plásmido pcDNA3.1 conteniendo la secuencia complementaria del gen SCN5A cDNA. Los oligonucleótidos para la mutagénesis fueron los siguientes:
P336LmutF: 5’-agctctgacgctgggacatgtctggagggcta-3’
P336LRev: 5’-gacatgtcccagcgtcagagctgttcccacaa-3’
I1660VmutF: 5’-cctgcctgccctcttcaacgtcgggctgct-3’
I1660VRev: 5’-gttgaagagggcaggcagggacatcatga-3’

Una vez realizada la mutagénesis de los plásmidos mutados se llevó a cabo la secuenciación para comprobar la presencia de las mutaciones y también la ausencia de mutaciones generadas por sustituciones de la enzima ADN polimerasa u otros errores técnicos de la duplicación.
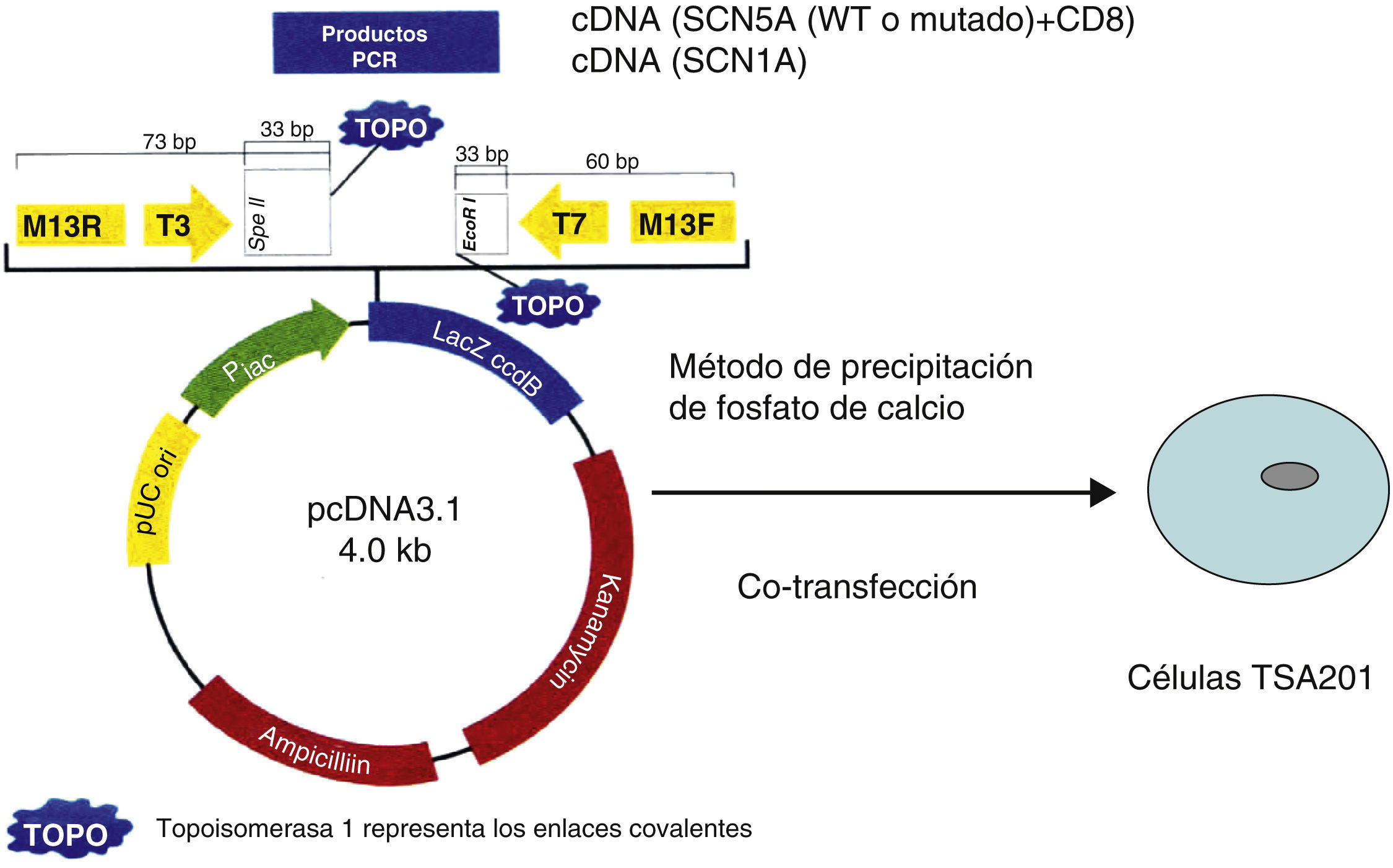
Transcripción in vitro y transfección celular mamíferaLos constructores genéticos fueron reclonados a partir de un vector original pcDNA3.1 (Invitrogen Carlbad, CA). Las mutaciones fueron construidas con el sistema de mutagénesis sitio-dirigida Gene TaylorTM (Invitro Corp) en el plásmido pcDNA3.1 conteniendo los oligonucleótidos apropiados. Los plásmidos mutados fueron secuenciados para comprobar la presencia de las mutaciones para estudio sin la presencia de sustituciones por errores técnicos. Las células humanas modificadas de riñón embrionarias (TSA201) fueron cotransfectadas con la misma cantidad de pcDNA usando el método de precipitación de fosfato de calcio (fig. 3). Las células crecieron en un ambiente de O2 (95%) y CO2 (5%) en placas de cultivo cubiertas de polilisina a una temperatura fisiológica de 37°C controlada mediante una incubadora (Medical Systems, Greenvale NY), por lo menos durante 2 días después de la transfección para su posterior estudio electrofisiológico. Cuando se estudió el efecto de incubación a bajas temperaturas se colocaron las cajas de cultivo a temperatura ambiente (TA) (20-22°C) adicionando un amortiguador de pH (HEPES 15mM) en el medio de cultivo.
Estudio biofísico de los canales de sodio SCN5A en las mutaciones P336L e I1660V y el marcador CD8+ en las células TSA201 mediante el método de precipitación por calcio.")
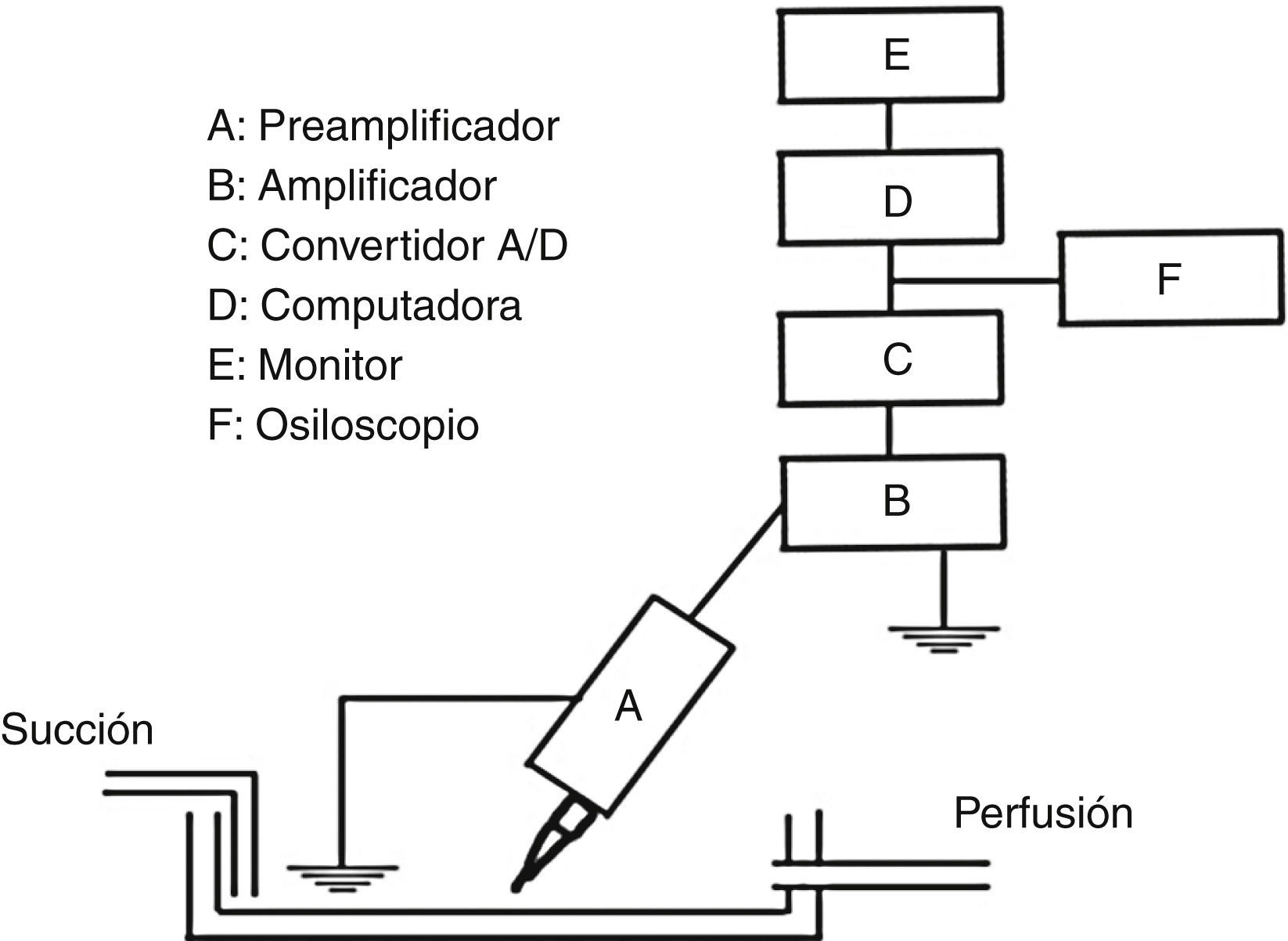
Para la realización de los estudios electrofisiológicos, el registro de los canales de sodio se determinó mediante la técnica de «fijación de voltaje en la configuración de célula completa» (fig. 4)6.
 y posteriormente se conectaron a un amplificador (B). Las corrientes iónicas de desplegaron para su constante visualización en la pantalla del ordenador como osciloscopio (E y F) y se digitalizaron mediante un convertidor analógico digital A/D (C), y los registros se almacenaron en un ordenador PC (D).")
Esquema del dispositivo experimental para obtener el registro de la INa+. Las células TSA201 se depositaron en una caja de Petri que permite la perfusión continua con las soluciones externas previamente burbujeadas con O2 al 100% y manteniendo el volumen constante por medio de una bomba de succión. Las micropipetas se acoplaron a un preamplificador (A) y posteriormente se conectaron a un amplificador (B). Las corrientes iónicas de desplegaron para su constante visualización en la pantalla del ordenador como osciloscopio (E y F) y se digitalizaron mediante un convertidor analógico digital A/D (C), y los registros se almacenaron en un ordenador PC (D).
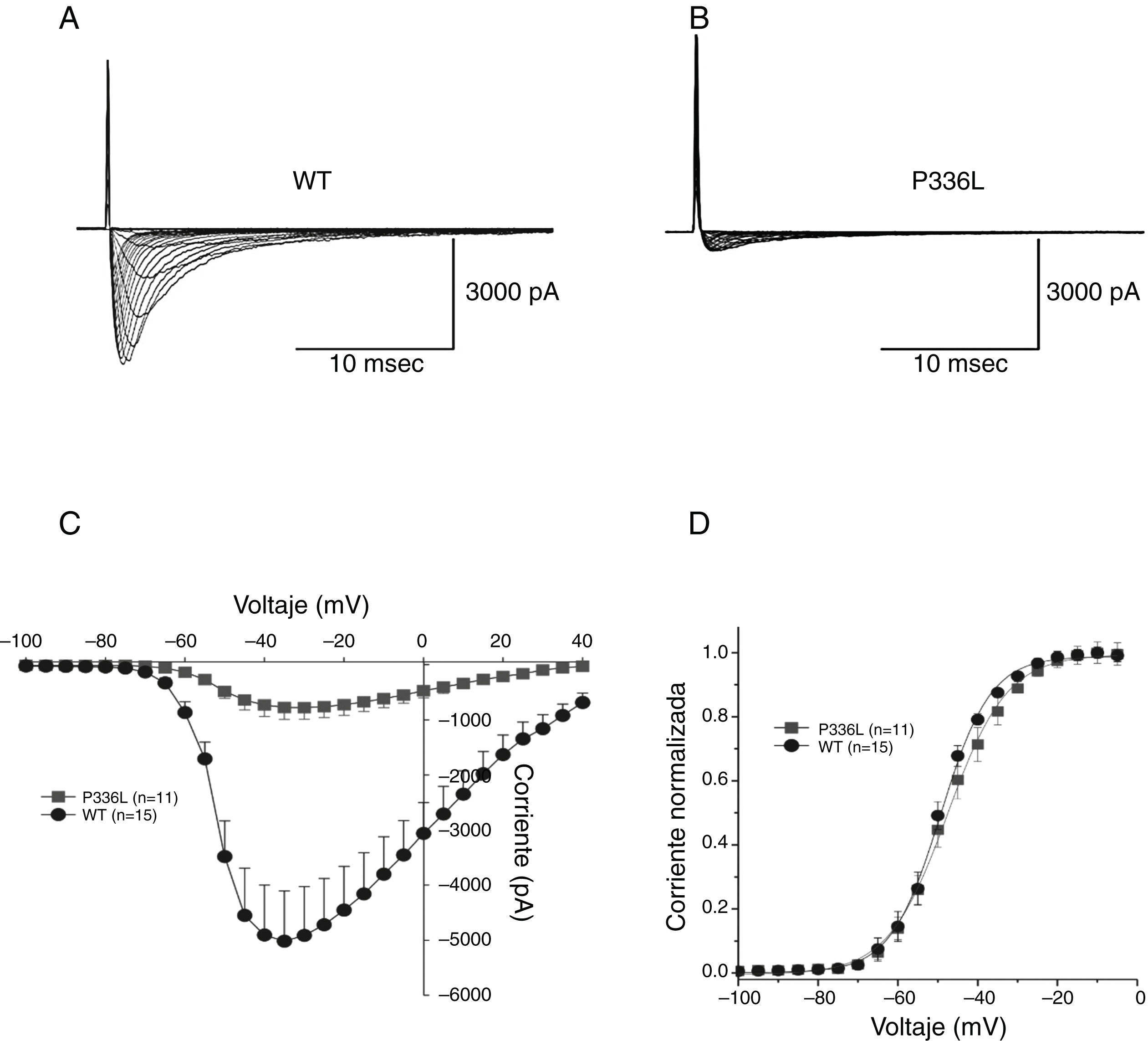
Los registros de la INa+ se obtuvieron mediante la aplicación de pulsos despolarizantes en incrementos de +5mV a partir de un potencial de reposo de −120mV. Con este protocolo se obtuvieron las corrientes de los canales silvestres de sodio (wild type, WT por sus siglas en inglés) y se compararon con los canales mutados P336L (figs. 5A y 5B). De los registros de la INa+ se muestra primero la relación corriente-voltaje de los canales WT y P336L. Al analizar la curva corriente-voltaje que muestra la densidad de INa+ como una medida de la magnitud de expresión de los canales de sodio por el gen SCN5A, se observó que la magnitud de la corriente P336L fue reducida de manera significativa comparada con la corriente WT. De manera adicional no se observó ningún cambio en el umbral de activación; tampoco hubo cambios en el pico máximo de la curva de activación (fig. 5C). El análisis del ajuste de la curva de activación en el estado estacionario muestra una pequeña variación no significativa del punto medio de activación entre los canales WT (49.3+0.27mV, n=15) y P336L (47.6+0.31mV, n=11), respectivamente (fig. 5D).
 y la mutación P336L (panel B) en la célula transfectada TSA201. Los registros se obtuvieron a partir de un potencial de prueba entre −100 y 0mV en incrementos de +5mV a partir de un potencial de mantenimiento de −120mV. Panel C: se muestra la relación corriente-voltaje para los canales WT (n=11) y P336L (n=15), mostrando una reducción en la corriente de sodio P336L. Panel D: gráfica que muestra la relación de la activación en el estado estacionario para WT y P336L. Panel C: la conductabilidad fue determinada usando la relación de la corriente con respecto al potencial electromotriz para cada célula. Los datos fueron normalizados y graficados al potencial de prueba. Modificada con permiso de Cordeiro et al.3.")
Efectos de la mutación P336L sobre la activación de la INa+. Se muestran los registros de la corriente de sodio para WT (panel A) y la mutación P336L (panel B) en la célula transfectada TSA201. Los registros se obtuvieron a partir de un potencial de prueba entre −100 y 0mV en incrementos de +5mV a partir de un potencial de mantenimiento de −120mV. Panel C: se muestra la relación corriente-voltaje para los canales WT (n=11) y P336L (n=15), mostrando una reducción en la corriente de sodio P336L. Panel D: gráfica que muestra la relación de la activación en el estado estacionario para WT y P336L. Panel C: la conductabilidad fue determinada usando la relación de la corriente con respecto al potencial electromotriz para cada célula. Los datos fueron normalizados y graficados al potencial de prueba. Modificada con permiso de Cordeiro et al.3.
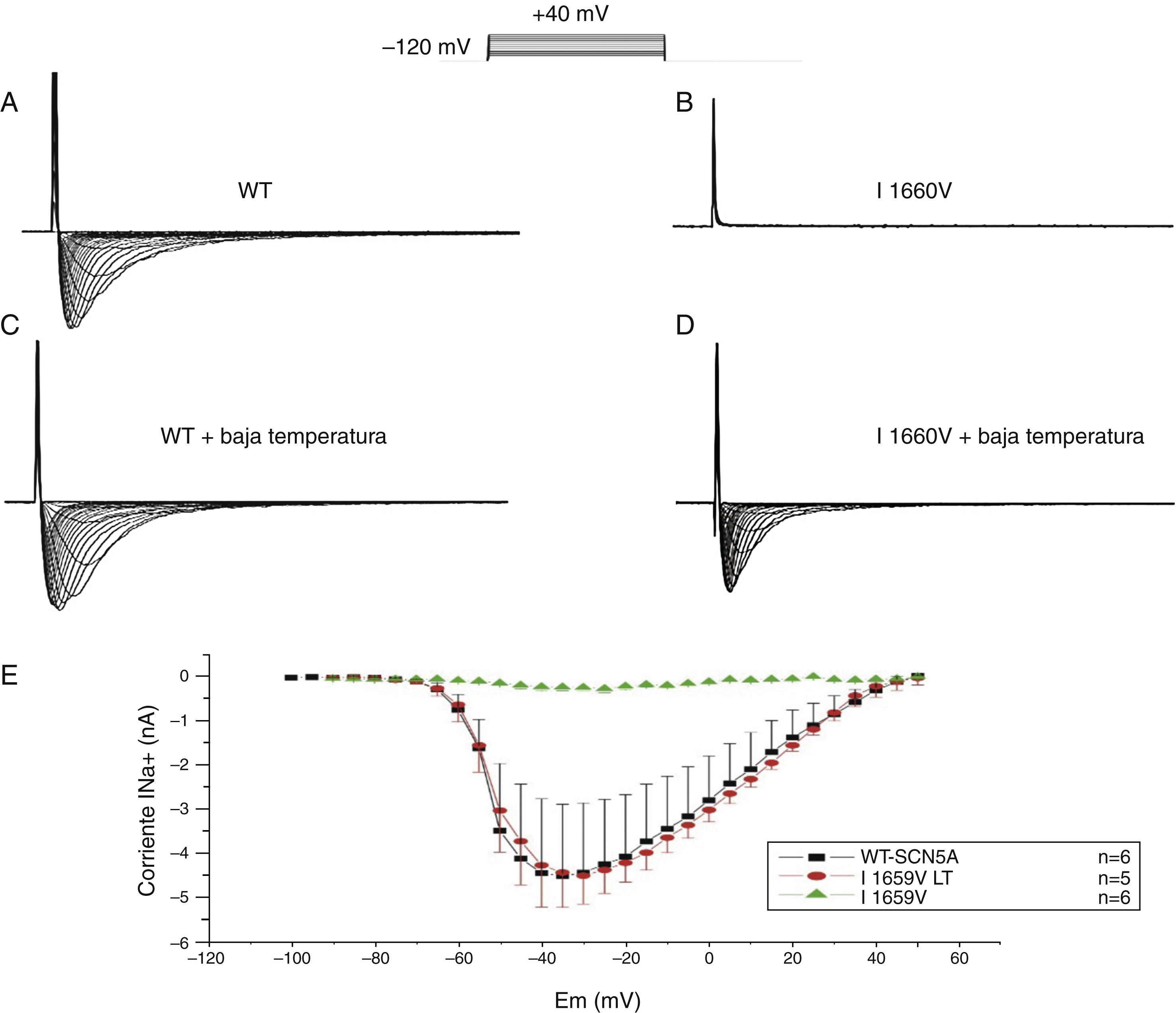
Posteriormente expresamos los canales I1660V en las células TSA201 y comparamos la relación corriente-voltaje con los canales WT. Las corrientes de sodio WT y las mutantes I1660V se obtuvieron mediante la aplicación del protocolo mostrado en las fig. 6A y 6B. Al realizar la curva corriente-voltaje se muestra que existe una alta disminución en la expresión en los canales de sodio I1660V (fig. 6E).
 y la mutación I1660V (B) en células TSA201 transfectadas. Las corrientes de sodio se obtuvieron a potenciales de prueba entre −100 a 0mV en incrementos de +5mV a partir de un potencial de mantenimiento de −120mV. Se realizó la incubación de las células TSA201 transfectadas a temperatura ambiente durante 48h para rescatar la corriente de sodio I1660V (D) y en la corriente de sodio WT no se observa ningún efecto por la incubación a baja temperatura (LT) (C). E: gráfica de la relación corriente-voltaje para los canales WT, la mutación I1660V y la incubación a temperatura ambiente de la mutación I1660V que muestra un rescate de la corriente de sodio. Modificada con permiso de Cordeiro et al.3.")
Efecto de la temperatura sobre los canales I1660V. Registros de la corriente de sodio para los canales WT (A) y la mutación I1660V (B) en células TSA201 transfectadas. Las corrientes de sodio se obtuvieron a potenciales de prueba entre −100 a 0mV en incrementos de +5mV a partir de un potencial de mantenimiento de −120mV. Se realizó la incubación de las células TSA201 transfectadas a temperatura ambiente durante 48h para rescatar la corriente de sodio I1660V (D) y en la corriente de sodio WT no se observa ningún efecto por la incubación a baja temperatura (LT) (C). E: gráfica de la relación corriente-voltaje para los canales WT, la mutación I1660V y la incubación a temperatura ambiente de la mutación I1660V que muestra un rescate de la corriente de sodio. Modificada con permiso de Cordeiro et al.3.
Se ha reportado que los canales mutados no funcionales podrían presentar un rescate, es decir, podrían recuperar los niveles normales de densidad de corriente por medio de varios métodos incluyendo la incubación a bajas temperaturas o un rescate mediante farmacología7–9. En la figura 6D se observa el grado de recuperación de los canales de sodio I1660V mediante la incubación de las células a TA (20-22°C).
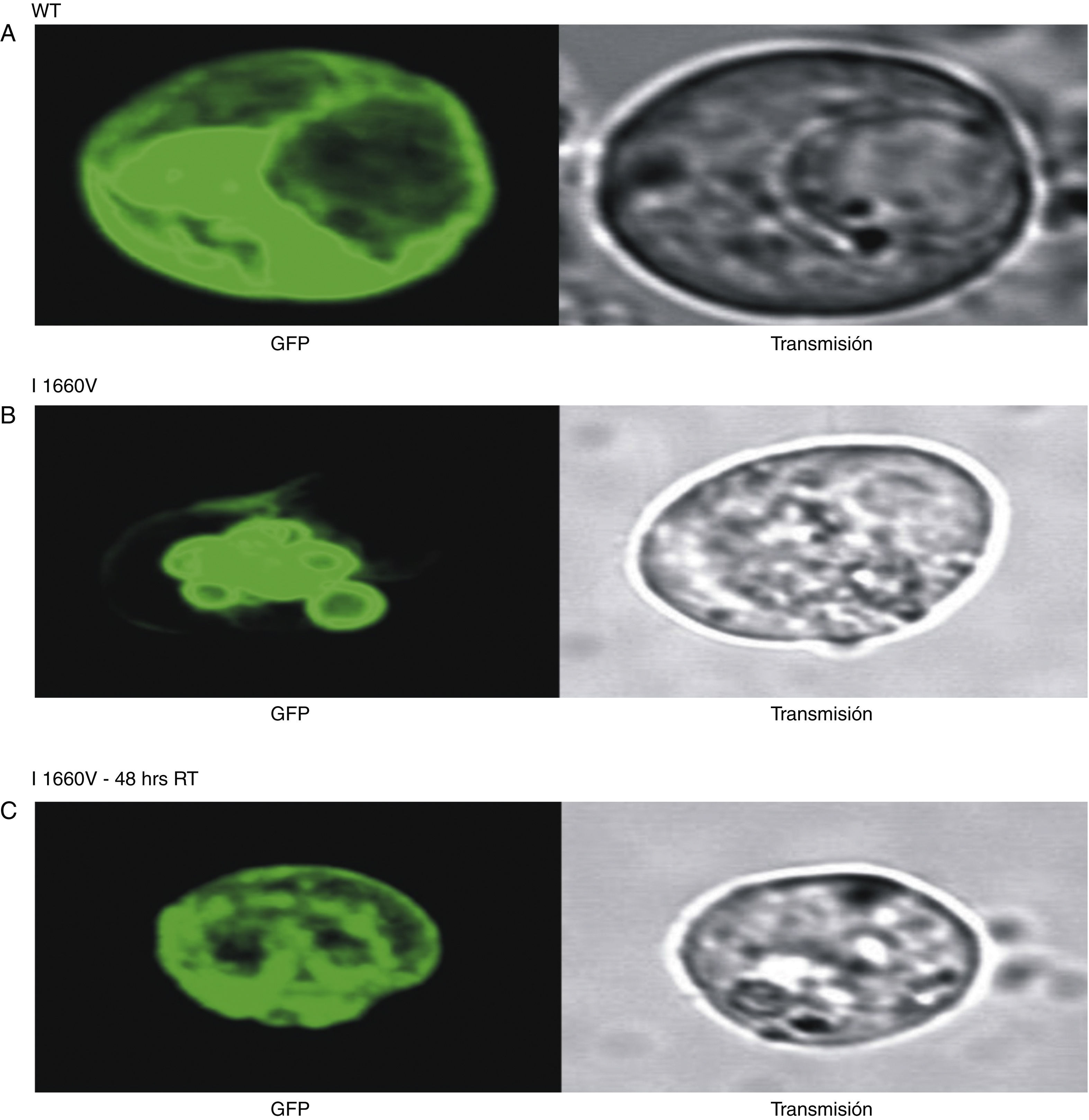
Estudios de localización de los canales de sodio conjugados con proteína verde fluorescenteLa disminución de la corriente de sodio por los canales de sodio I1660V puede deberse a un defecto en el tráfico intracelular hacia la membrana celular. Una manera de estudiar el rescate es mediante la disminución de la temperatura de incubación y mediante el uso de fármacos que permiten el rescate de los canales7–9. Para confirmar esta hipótesis, nosotros llevamos a cabo experimentos mediante microscopia confocal realizando la formación de constructos genéticos de los genes de los canales de sodio WT e I1660V adicionando un gen reportero fluorescente llamado proteína verde fluorescente, y de esta manera, estudiar los patrones de localización del canal fluorescente (fig. 7, WT). Los experimentos se realizaron en un microscopio confocal de búsqueda láser FluoView Olympus y las imágenes fueron adquiridas con el programa FluoView en un ordenador. Las células TSA201 marcadas con la proteína verde fluorescente fueron analizadas en la configuración XYZ, previamente descrita10.
 en el interior y la periferia de la membrana plasmática lo que sugiere el tráfico normal de los canales del interior a la membrana celular. En contraste, en el panel B se muestra el canal mutado I1660V con fluorescencia solo en la región perinuclear de la célula, evidencia que sugiere anormalidad en el tráfico del canal hacia la membrana incubados a 37°C. En el panel C se evidencia el rescate de los canales hacia la membrana plasmática de los canales mutados I1660V mediante incubación a temperatura ambiente. Fuente: con permiso de Cordeiro et al.3.")
Efecto de la temperatura sobre el tráfico del canal mutado I1660V. Imágenes XYZ de microscopia confocal fluorescente mostrando la localización de los canales de sodio en las células TSA201. Panel A: se muestran los canales WT conjugados con la proteína verde fluorescente (green fluorescent protein, GFP por sus siglas en inglés) en el interior y la periferia de la membrana plasmática lo que sugiere el tráfico normal de los canales del interior a la membrana celular. En contraste, en el panel B se muestra el canal mutado I1660V con fluorescencia solo en la región perinuclear de la célula, evidencia que sugiere anormalidad en el tráfico del canal hacia la membrana incubados a 37°C. En el panel C se evidencia el rescate de los canales hacia la membrana plasmática de los canales mutados I1660V mediante incubación a temperatura ambiente. Fuente: con permiso de Cordeiro et al.3.
Las células TSA201 con los canales de sodio WT e I1660V fueron incubados a TA durante 48h previo al registro de la corriente. La incubación a TA de los canales de sodio WT no afectó la magnitud de la corriente de sodio (fig. 6C), en cambio se observó un significativo rescate de los canales de sodio mutados I1660V incubados a TA a niveles similares a la corriente WT (fig. 6E). De manera opuesta, la incubación a TA de los canales de sodio P336L no produjo ningún rescate de la corriente (datos no mostrados).
La localización por fluorescencia de los canales de sodio WT mostró una distribución tanto en el centro como en la periferia de las células, sugiriendo que los canales son formados desde el interior y llevados hasta su incorporación a la membrana celular (fig. 7A). En contraste, los canales de sodio mutados I1660V presentaron fluorescencia solamente en el interior de la célula, sugiriendo que los canales traducidos permanecen atrapados en los organelos celulares (fig. 7B). De esta manera confirmamos que la incubación a TA incrementó la localización de los canales de sodio I1660V en la membrana de las células transfectadas. En las figura 7C y 7D se muestra la localización confocal de los canales de sodio I1660V incubados a TA, mostrando fluorescencia en la parte interna y en la periferia de la célula, lo que sugiere que el tráfico de los canales es favorecido mediante la incubación a bajas temperaturas.
Correlación clínica con el síndrome de BrugadaEstá bien establecido que una reducción en la corriente de sodio cardiaca es responsable de la manifestación del SBr. La presencia de mutaciones en el gen SCN5A puede producir esta disminución y generar el fenotipo del SBr. Sin embargo, solamente al 20% de los pacientes con SBr se les ha detectado mutaciones del canal de sodio; esto sugiere que el síndrome es genéticamente heterogéneo11. En el presente estudio, el análisis biofísico del canal mutado I1660V produjo una disminución de la corriente de sodio expresada en las células TSA201. En teoría, un individuo que presenta una mutación produciría un 50% de pérdida de la corriente de sodio. Sin embargo, los datos clínicos de las hijas del caso índice que presentaron una mutación muestran que ninguna de ellas exhibió cambios en la conducción, sin elevación del segmento ST y solo una de ellas presentó una ligera prolongación del intervalo PR del ECG12.
Los canales P336L produjeron una reducción apreciable de la magnitud de la corriente de sodio (aproximadamente el 80% comparada con la WT). Sin embargo, la paciente con esta mutación presentó un ECG normal. Observaciones similares fueron descritas en un reciente estudio13. Identificaron a un paciente con defectos severos de la conducción y anormalidades en el ECG. El análisis genético mostró que los individuos presentaron 2 mutaciones en el canal de sodio cardiaco en diferentes alelos. Una mutación fue heredada por vía materna y la otra por vía paterna. Es interesante que tanto el padre como la madre no mostraron anormalidades en el ECG, sugiriendo que las mutaciones mostraron una baja penetrancia para inducir un efecto13.
Existen varios factores que pueden influir en el grado de penetrancia de un canal de sodio mutado. Por ejemplo, los defectos genéticos son heredados al 50% en los individuos descendientes, y tanto hombres como mujeres heredan igualmente el defecto genético pero no todos desarrollarán la enfermedad. No obstante, la transmisión genética de manera equitativa de la mutación podría causar un fenotipo clínico de SBr de 8 a 10 veces de mayor prevalencia en los hombres que en las mujeres. Esta relación de resultados por género podría explicarse debido a que los descendientes femeninos no exhiben un ECG anormal parecido al SBr. Existen también variaciones electrocardiográficas importantes como las descritas en algunos pacientes con SBr que presentan cambios dependientes de la frecuencia de estimulación durante la fibrilación auricular o durante un episodio de muerte súbita abortada14,15.
Parte de este material fue publicado originalmente en inglés (Cordeiro JM, Barajas-Martinez H, Hong K, et al. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 2006;114: 2026-33) y es reproducido con autorización y para fines exclusivamente didácticos.