


Hombre de 51 años admitido en el hospital por presentar palpitaciones y mareos de 2h de evolución. El electrocardiograma demostró taquicardia regular de QRS ancho y frecuencia cardíaca de 250lpm, con eje superior y morfología de bloqueo completo de rama izquierda sin descompensación hemodinámica. Se administraron dosis de carga y mantenimiento con amiodarona, revirtiendo a ritmo sinusal. El estudio electrofisiológico demostró el origen ventricular de la taquicardia y su inducibilidad. En la angiografía coronaria no se observaron lesiones significativas en los vasos epicárdicos. Se realizó un ecocardiograma Doppler que presentó cavidades con diámetros y función sistólica y diastólica dentro de los parámetros normales. Ante la sospecha de enfermedad estructural miocárdica se llevó a cabo una resonancia magnética cardíaca contrastada con realce tardío que demostró alteración estructural del ventrículo derecho con incremento de la trabeculación e infiltración fibrograsa parietal y deterioro moderado de su función sistólica, y deterioro leve de la función sistólica del ventrículo izquierdo, lo cual permitió realizar el diagnóstico de miocardiopatía arritmogénica del ventrículo derecho por presentar 2 criterios mayores. Se decidió implantar un cardiodesfibrilador automático, para prevenir la muerte súbita. El paciente evolucionó de manera favorable y fue dado de alta.
A 51-year-old man was admitted to this hospital because of palpitations and a feeling of dizziness for a period of 2h. The electrocardiogram revealed a regular wide-QRS complex tachycardia at a rate of 250 beats per minute, with superior axis and left bundle branch block morphology without hemodynamically decompensation, the patient was cardioverted to sinus rhythm after the administration of a loading and maintenance dose of amiodarone. The elechtrophysiological study showed the ventricular origin of the arrhythmia. In order to diagnose the etiology of the ventricular tachycardia we performed a coronary arteriography that showed normal epicardial vessels, thus ruling out coronary disease. Doppler echocardiography revealed systolic and diastolic functions of both left and right ventricles within normal parameters, and normal diameters as well. A cardiac magnetic resonance with late enhancement was done, showing structural abnormalities of the right ventricle wall with moderate impairment of the ejection fraction, and a mild dysfunction of the left ventricle. The diagnosis of arrhythmogenic right ventricular cardiomyopathy was performed as 2 major Task Force criteria were met. We implanted an automatic cardioverter defibrillator as a prophylactic measure. The patient was discharged without complications.
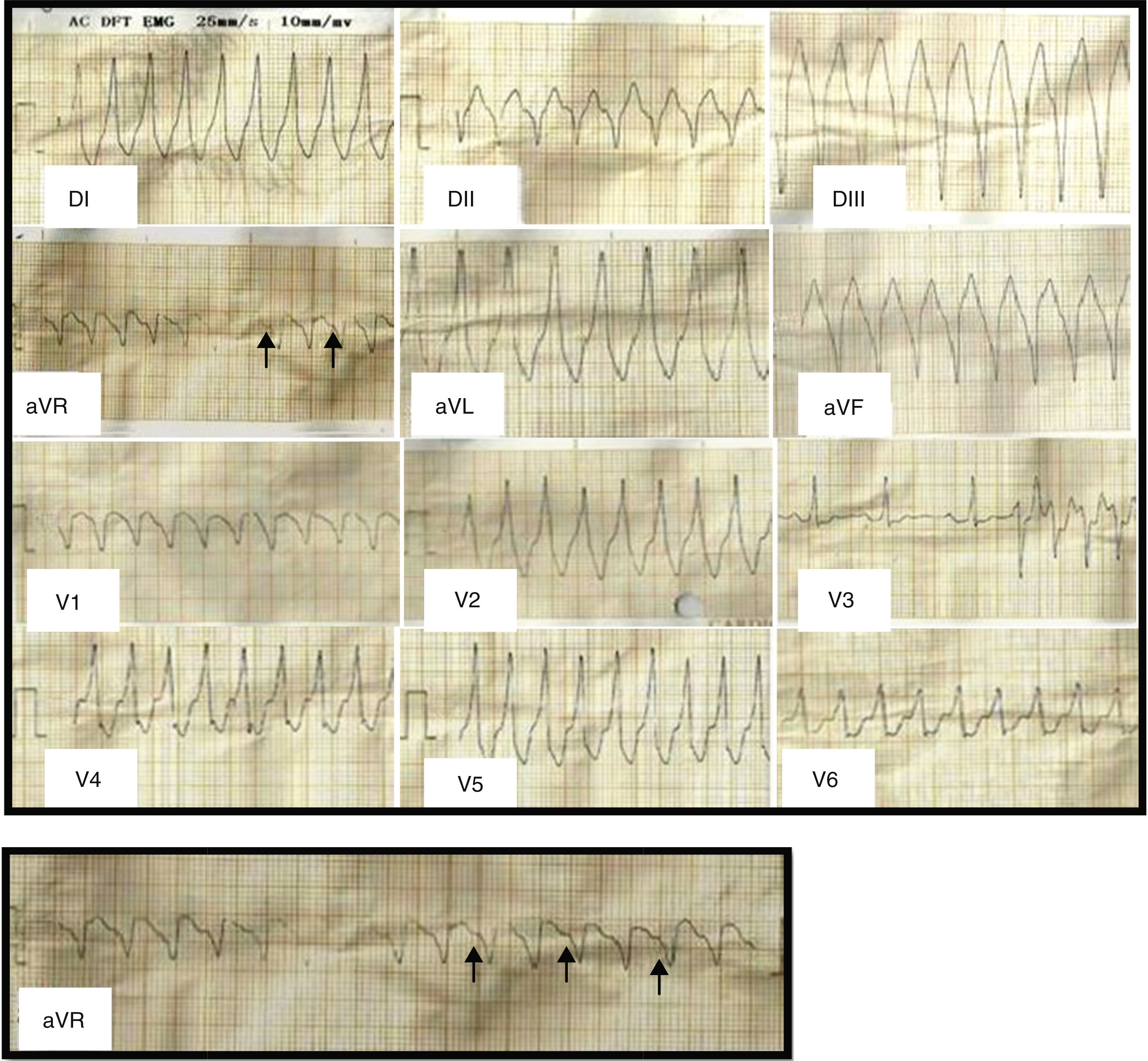
Hombre de 51 años de edad, fumador de 10 cigarrillos/día. Acudió al departamento de emergencias por presentar palpitaciones y mareos de 2h de evolución. Refirió haber presentado episodios similares hace 3 meses, pero no acudió a consulta. También se le realizaron estudios cardiológicos anuales y los electrocardiogramas (ECG) siempre fueron normales y no contaba con antecedentes heredo-familiares cardiovasculares de relevancia. En el momento de la evaluación presentaba palpitaciones y se encontraba hemodinámicamente estable, con tensión arterial de 100/60mmHg. En el ECG (fig. 1) se registró taquicardia de QRS ancho regular (>120mseg), con una frecuencia cardíaca de 250lpm, eje eléctrico de −35°. Se analizaron los criterios de Brugada para taquiarritmia de QRS ancho y, aunque de manera no concluyente, fue calificado para taquicardia ventricular (TV) por el tercer criterio al observarse en alguna derivación (aVR) disociación auriculoventricular1. Debido a que no había descompensación hemodinámica y al ser la arritmia bien tolerada por el paciente, se realizó masaje del seno carotideo por breves segundos; esta acción no terminó con la arritmia. Se administró carga de amiodarona con 5mg/kg en 30min y se mantuvo con 15mg/kg/día, lográndose la reversión de la arritmia a ritmo sinusal en aproximadamente 30min; posteriormente se rotó por atenolol a 25mg b.i.d. Como particularidad, en dicho ECG se observa onda T negativa en V1 y elevación del punto J en cara anterior de 0.5mm (fig. 2).
 ECG de ingreso: taquicardia de QRS ancho regular (>120 mseg), con una FC de 250lpm y eje superior. Las flechas señalan las ondas P disociadas. B) aVR magnificada. Las flechas señalan las ondas P disociadas.")

Al ingreso, los estudios de laboratorio mostraron: hematocrito de 45%, creatinina 1.43mg/dL, sodio 135mEq/L, potasio 4.12mEq/L, calcemia 9mg/dL, calcio iónico 4.5mg/dL, fosfatemia 3.7mg/dL, magnesemia 2.2mg/dL, TSH 2.43μU/ml, T4 libre 1.33ng/dL.
El ecocardiograma Doppler transtorácico mostró diámetro diastólico del ventrículo izquierdo (VI) de 50mm, diámetro sistólico del VI de 31mm, septum interventricular de 10mm, pared posterior de 9mm, diámetro diastólico del ventrículo derecho (VD) de 21mm, aurícula izquierda de 37mm, fracción de expulsión del VI de 64%, motilidad en reposo conservada sin alteraciones regionales, cavidades derechas normales con una motilidad del VD dentro de los parámetros normales.
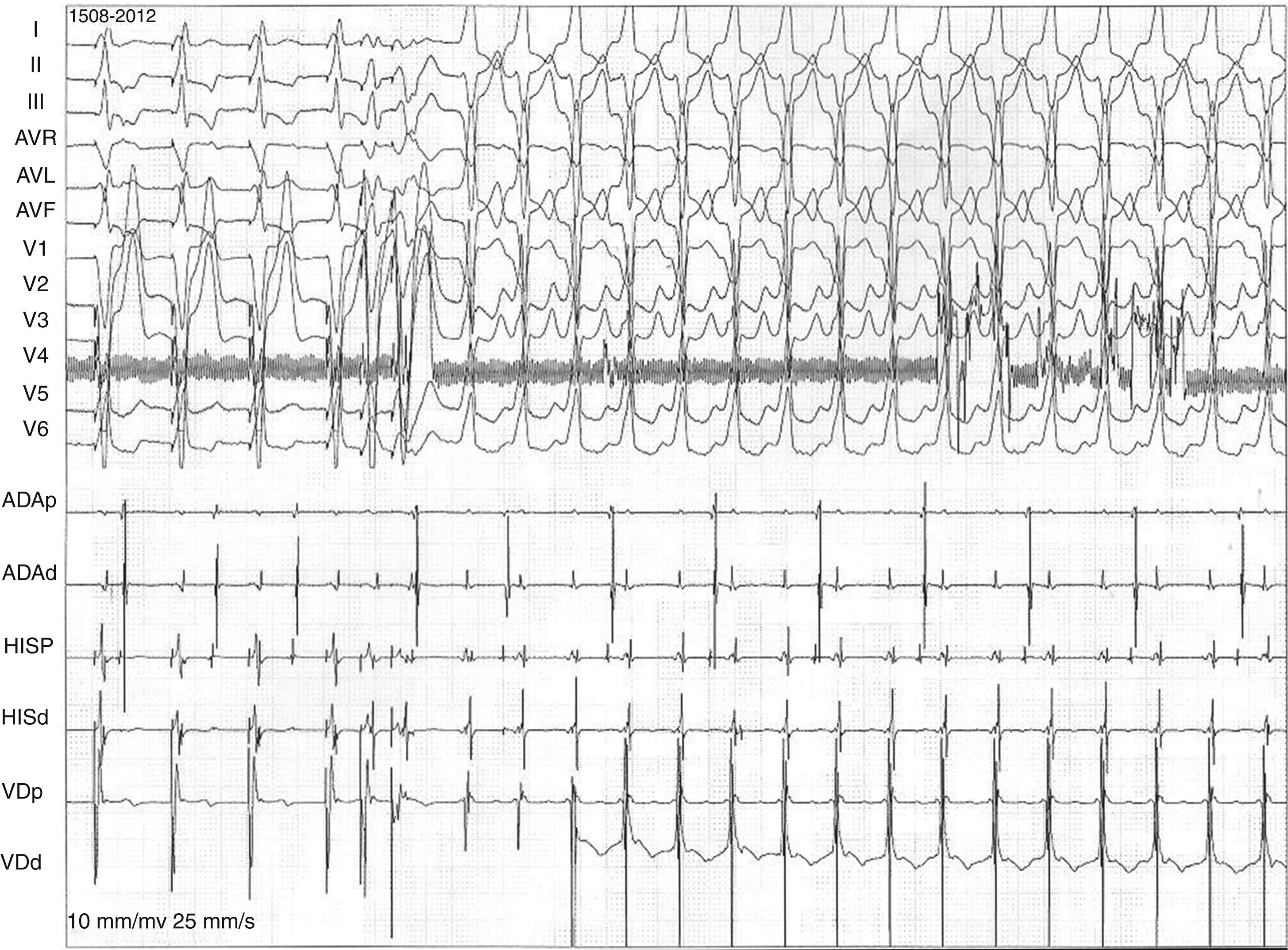
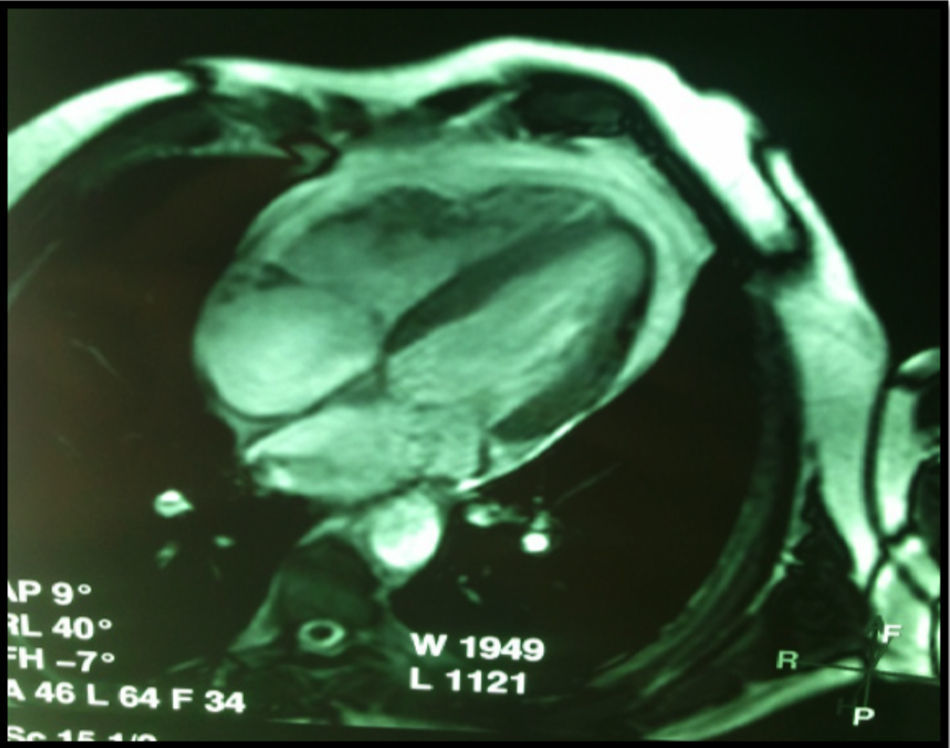
Se realizó un estudio electrofisiológico. Durante la estimulación programada desde ápex de VD con 600mseg se observó falta de conducción retrógrada, posteriormente se indujo TV de morfología idéntica a la clínica en reiteradas ocasiones con una longitud de ciclo de 412mseg con 2 extraestímulos (270 y 240mseg) sobre el ciclo basal de 600mseg. Conclusión: TV monomorfa, no sostenida e inducible en repetidas ocasiones. La inducibilidad reiterada con estimulación programada habla de mecanismo de reentrada como génesis de la TV (figs. 3 y 4). Como diagnósticos diferenciales se plantearon: 1) cardiopatía isquémica, 2) miocardiopatía/displasia arritmogénica del ventrículo derecho (MAVD), 3) TV idiopática del tracto de salida del ventrículo derecho y 4) miocarditis. Para descartar la cardiopatía isquémica se realizó una angiografía coronaria que mostró arterias coronarias epicárdicas sin lesiones significativas; la TV idiopática del tracto de salida del VD no es reentrante y por lo tanto no es fácilmente inducible, y se descartó por el comportamiento electrofisiológico. Para descartar los restantes diagnósticos, se realizó resonancia magnética nuclear cardíaca con gadolinio con realce tardío (fig. 5) la cual mostró cavidades derechas mínimamente dilatadas con volumen al final de diástole del VD de 129.3ml, volumen al final de sístole del VD de 79.2ml, con incremento de la trabeculación, pared libre adelgazada y de bordes irregulares; también se observó infiltración fibrograsa a nivel basal, con leve hipocinesia global del VD, con áreas de acinesia regional a nivel medioventricular, fracción de expulsión del VD del 38.7%, área de aurícula derecha de 20.2cm2, tenue presencia de realce tardío de contraste sobre la pared libre del VD a nivel basal, sin observarse realce tardío de contraste en el resto del miocardio. En cuanto al VI, el estudio mostró volumen al final de diástole del VI de 135ml, volumen al final de sístole del VI de 66.8ml, fracción de expulsión del VI del 50.5%, con mínima hipocinesia global sin trastornos regionales, área de aurícula izquierda de 21.8cm2, septum interventricular de 10.4mm, raíz de la aorta de 32.2mm y válvulas sin regurgitación significativa.


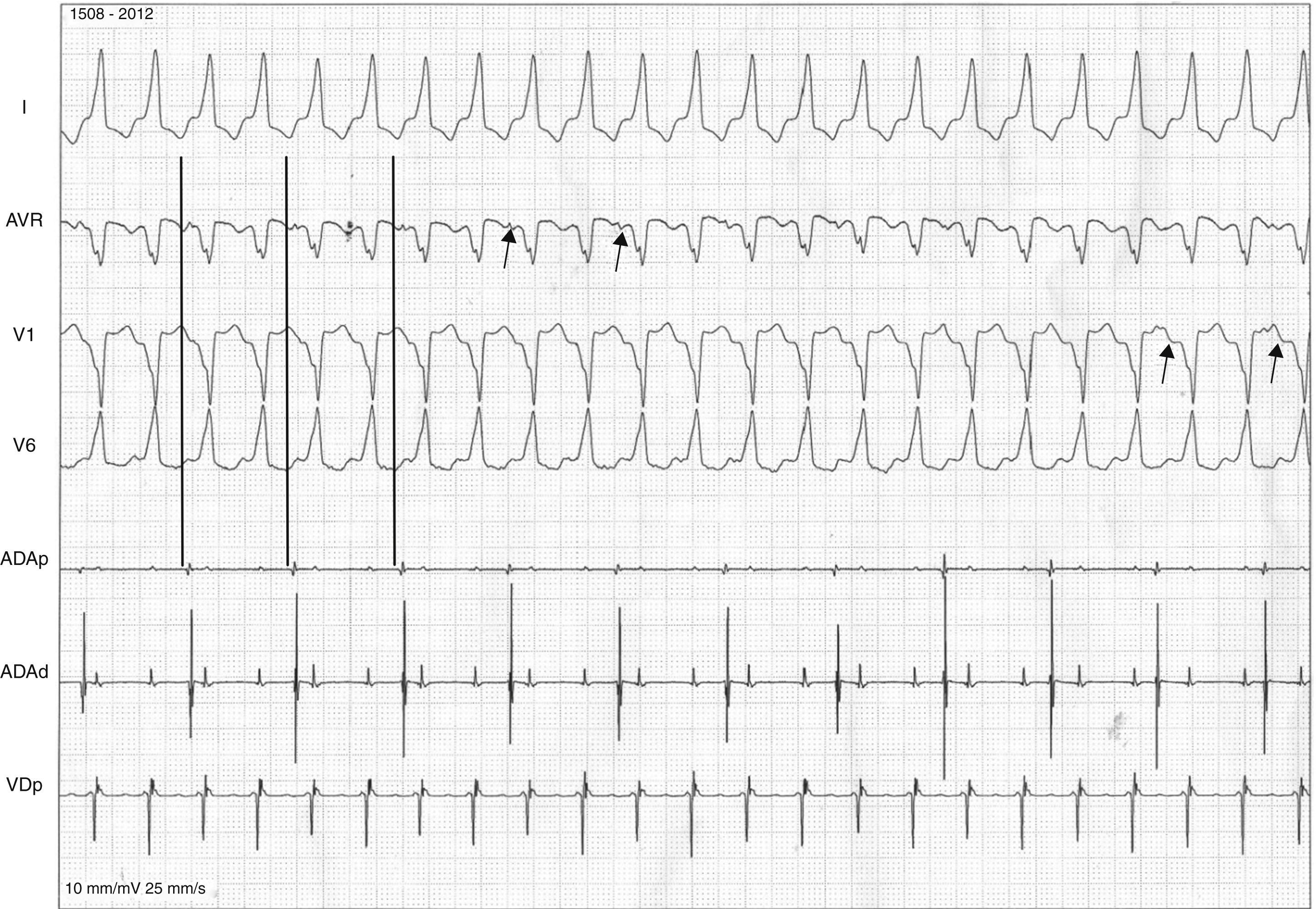
Estudio electrofisiológico donde se observa TV monomorfa inducible con conducción VA 2:1 que determina en el ECG de superficie la presencia de ondas P retrógradas con relación 2:1 con el QRS visibles principalmente en aVR y V1 coincidentes con los electrogramas endocavitarios de estimulación auricular. Las flechas y las líneas transversales señalan la disociación AV en los ECG de superficie, siendo más claro en aVR y en determinados pasajes en V1, no así en V6. Se puede observar claramente la aparición de una onda P cada 2 QRS.

De esta manera se llegó al diagnóstico de MAVD por presentar 2 criterios mayores según la nueva revisión de la Task Force2: TV sostenida con morfología de bloqueo completo de rama izquierda (BCRI), con eje superior y acinesia regional del VD con una FEVD menor del 40%. Por consiguiente se decidió la colocación de un cardiodesfibrilador implantable (CDI) y se indicó atenolol 25mg b.i.d.; el paciente evolucionó favorablemente.
DiscusiónEl concepto de miocardiopatía específica del VD se propuso por primera vez en 1977 por Fontaine et al., en una descripción de 6 pacientes con TV sostenida y dilatación del VD3. En el año 1982 en un artículo de Marcus et al. fue publicado el término «displasia arritmogénica del VD» en pacientes adultos con taquiarritmia ventricular con morfología de BCRI, anomalías del movimiento de la pared del VD y sustitución del miocardio del VD por tejido adiposo y fibroso4. Las características electrocardiográficas, incluida la onda épsilon, fueron descritas por primera vez en 19845. Sin embargo, no fue reconocida formalmente como una entidad diferenciada sino hasta 1994, tras la publicación de los criterios diagnósticos de la Task Force de la Organización Mundial de la Salud6. Desde entonces, la displasia arritmogénica del VD ha sido reconocida como una importante causa de muerte súbita en personas jóvenes y particularmente en atletas7.
Es una enfermedad del músculo cardíaco caracterizada por pérdida de miocardiocitos y sustitución de los mismos por tejido fibroso o fibroadiposo, que puede dar lugar a arritmias, muerte súbita e insuficiencia cardíaca. Con frecuencia se hereda la forma autosómica dominante, aunque existen formas recesivas (enfermedad de Naxos y el síndrome de Carvajal) asociadas a un fenotipo cutáneo. Se caracteriza por mutaciones en genes codificadores de proteínas que intervienen en la unión célula-célula2,8.
El reemplazo por tejido fibroadiposo es progresivo, comienza desde el epicardio o miocardio medio, se extiende transmuralmente y conduce progresivamente al afinamiento y a la formación de aneurismas parietales, que típicamente se localizan en la pared inferior, apical e infundibular del VD, comúnmente llamado «triángulo de la displasia»9. Es común la evolución hacia un compromiso difuso del VD y afección del VI que afecta la pared posterior, siendo también reconocida la forma con predominio del VI10,11.
Es difícil estimar la prevalencia en la población general debido a las dificultades diagnósticas. En Europa, estudios realizados indican una prevalencia de entre 0.6 y 4.4‰ y es causa de muerte súbita cardíaca en el 11-27% de los casos de pacientes menores de 35 años2.
Afecta más a hombres que a mujeres en una relación de 3:1 y se hace clínicamente manifiesta, en general, entre la segunda y la cuarta década de vida, siendo poco frecuente su presentación antes de la pubertad o después de los 60 años9; aunque pudiese estar subdiagnosticada en este último grupo etario por baja sospecha.
La presentación clínica es variable; puede presentarse con palpitaciones, síncope, muerte súbita abortada, siendo menos frecuentes la insuficiencia cardíaca derecha o la biventricular que simulen una miocardiopatía dilatada9.
En la actualidad, para diagnosticarla, se emplean los criterios modificados en el año 2010 por la Task Force, los cuales se dividen en distintas categorías que incluyen: 1) disfunción y alteraciones estructurales generales o regionales, 2) caracterización del tejido de la pared, 3) anomalías de la repolarización, 4) anomalías de la despolarización/conducción, 5) arritmias y 6) antecedentes familiares, que a su vez se subdividen en mayores y menores; de estas emerge el diagnóstico definitivo con 2 criterios mayores, uno mayor más 2 menores, o 4 criterios menores de diferentes grupos2,8,12.
El tratamiento consiste principalmente en la prevención de la muerte súbita12. La actividad deportiva se asocia a un incremento de 5 veces del riesgo de muerte súbita en pacientes con MAVD y debe ser restringida13. En pacientes que presentan episodios de arritmias sin descompensación hemodinámica, como TV sostenida o no sostenida, los β-bloqueadores o antiarrítmicos de clase iii (sotalol y amiodarona), empleados solos o en combinación, han demostrado ser los agentes más efectivos, con un bajo efecto proarritmogénico14. El tratamiento con CDI está indicado en pacientes con paro cardíaco, síncope o descompensación hemodinámica después de una TV, a pesar del tratamiento con fármacos antiarrítmicos15. En la actualidad no se recomienda el implante profiláctico de CDI en pacientes asintomáticos o con manifestaciones mínimas de la enfermedad debido al pronóstico general favorable y al elevado riesgo de complicaciones relacionadas con el dispositivo o electrodo durante el seguimiento a medio plazo. Una excepción la constituyen los pacientes con disfunción ventricular derecha grave y temprana, o en estadio avanzado de la enfermedad con compromiso biventricular, quienes probablemente presenten un elevado riesgo de muerte súbita independientemente de haber padecido o no eventos arrítmicos previos15. En aquellos pacientes con múltiples factores de riesgo, historia familiar de muerte súbita o TV/fibrilación ventricular inducida por medio de un estudio electrofisiológico, es controvertido y requiere del estudio individualizado del caso16. La resección con catéter de radiofrecuencia del foco arritmogénico de TV tiene una tasa de éxito agudo del 60-90%, pero debido a la naturaleza progresiva de la enfermedad, las recidivas son frecuentes, con una tasa de TV recurrente de hasta un 90% a los 3 años, por lo que su uso es limitado17–19. En los casos de insuficiencia cardíaca refractaria o arritmias ventriculares intratables, el trasplante cardíaco constituye una opción terapéutica final20.
En el caso de nuestro paciente, la manifestación inicial de la MAVD consistió en palpitaciones y se presentó como TV sostenida sin descompensación hemodinámica, con morfología de BCRI y eje superior, sin onda épsilon evidenciable en el ECG. En el ecocardiograma Doppler no presentaba anomalías particulares, pero debido a las limitaciones del método para evaluar el VD y por la justificada sospecha clínica, se indicó realizar una resonancia magnética nuclear con realce tardío. La misma presenta claras ventajas sobre la ecocardiografía entre las cuales cabe mencionar la visualización tridimensional del corazón, la cuantificación de los volúmenes del VD y el VI, la excelente resolución espacial y la caracterización del tejido. Por otro lado, la captación tardía de contraste con gadolinio y una infiltración fibroadiposa del miocardio, en especial en el VI, pueden ser útiles para identificar a los individuos afectados2,21,22. Nuestro paciente presentaba deterioro moderado de la función sistólica del VD con trastornos globales y segmentarios de la motilidad, y evidencia de miocardiopatía estructural por afinamiento parietal y depósito fibroadiposo. Asimismo, presentaba compromiso leve del VI el cual, según las diferentes publicaciones, se encuentra involucrado en relación directa con la progresión de la enfermedad asociado a falla biventricular10,12.
De esta manera se realizó el diagnóstico de MAVD por presentar el paciente 2 criterios mayores. Se decidió la colocación de un CDI por haber tenido episodios de TV a su ingreso en el hospital e inducidos durante el estudio electrofisiológico, aunque sin compromiso hemodinámico, asociados a falla biventricular incipiente con predominio ventricular derecho, es decir, como prevención primaria.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.