









La Aniridia: Actualización en Diagnóstico, Complicaciones y Tratamiento
Más datosLa aniridia es una enfermedad panocular caracterizada por hipoplasia iridiana, habitualmente acompañada de otras manifestaciones oculares, con gran variabilidad y solapamiento clínico con otras anomalías del segmento anterior y posterior. Esta revisión se centra en las características genéticas de esta enfermedad autosómica dominante causada por mutaciones en el gen PAX6. El espectro mutacional es muy amplio, con más de 700 mutaciones descritas hasta la fecha, siendo las mutaciones que causan un codón de parada prematuro, las más frecuentes. Los recientes avances en las técnicas de secuenciación masiva (NGS) han aumentado el rendimiento diagnóstico en la aniridia, y han contribuido a dilucidar nuevos mecanismos que conducen a la haploinsuficiencia de PAX6. En esta revisión, actualizamos los protocolos de diagnóstico genético incluyendo estas nuevas técnicas de NGS para realizar estudios con mayor coste-efectividad y rapidez. Su uso abre la posibilidad de estudiar simultáneamente las variaciones puntuales y estructurales tanto en PAX6 como en otros genes de diagnóstico diferencial con otras enfermedades solapantes. Algunos pacientes con fenotipos atípicos pueden presentar mutaciones en FOXC1 y PITX2, genes asociados a un amplio espectro de disgenesias del segmento anterior, o en ITPR1, responsable de una forma distintiva de aplasia circumpapilar del iris presente en el síndrome de Gillespie, entre otros. Dado que la aniridia se puede presentar asociada a formas sindrómicas, como el síndrome de WAGR causado por deleciones contiguas de PAX6 y WT1, los estudios genéticos son cruciales para realizar un correcto diagnóstico y manejo clínico de los pacientes, además de permitir diagnósticos prenatales y preimplantacionales en las familias.
Aniridia is a panocular disease characterized by iris hypoplasia, accompanied by other ocular manifestations, with a high clinical variability and overlapping with different abnormalities of the anterior and posterior segment. This review focuses on the genetic features of this autosomal dominant pathology, which is caused by the haploinsufficiency of the PAX6 gene. Mutations causing premature stop codons are the most frequent among the wider mutational spectrum of PAX6, with more than 600 different mutations identified so far. Recent advances in next-generation sequencing (NGS) have increased the diagnostic yield in aniridia and contributed to elucidate new etiopathogenic mechanisms leading to PAX6 haploinsufficiency. Here, we also update good practices and recommendations to improve genetic testing and clinical management of aniridia using more cost-effective NGS analysis. Those new approaches also allow studying simultaneously both structural variants and point-mutations in PAX6 as well as other genes for differential diagnosis, simultaneously. Some patients with atypical phenotypes might present mutations in FOXC1 and PITX2, both genes causing a wide spectrum of anterior segment dysgenesis, or in ITPR1, which is responsible for a distinctive form of circumpupillary iris aplasia present in Gillespie syndrome, or other mutations in minor genes. Since aniridia can also associate extraocular anomalies, as it occurs in carriers of PAX6 and WT1 microdeletions leading to WAGR syndrome, genetic studies are crucial to assure a correct diagnosis and clinical management, besides allowing prenatal and preimplantational genetic testing in families.
La aniridia congénita (MIM #106210) es una enfermedad panocular caracterizada por el desarrollo anómalo del iris y de la fóvea. En general, la aniridia presenta una alta variabilidad clínica con distintas manifestaciones en el segmento anterior y posterior del ojo1. Los pacientes pueden manifestar fotofobia, nistagmo, estrabismo y un déficit visual que puede progresar a la ceguera legal. Aunque suele presentarse de forma aislada, la aniridia aparece en un tercio de los pacientes dentro un contexto sindrómico, siendo el síndrome de WAGR (MIM #194072) la forma más frecuente1.
La aniridia es una enfermedad monogénica con un patrón de herencia autosómico dominante que está causada mayoritariamente por mutaciones heterocigotas en el gen PAX6, un gen clave en la morfogénesis ocular. Los estudios moleculares en la aniridia son una herramienta diagnóstica imprescindible para la confirmación clínica, el manejo del paciente y el asesoramiento familiar2. En los últimos años, los avances tecnológicos en el campo de la medicina genómica han contribuido a la descripción de nuevos mecanismos moleculares en la etiopatogenia de la aniridia que implican variantes en regiones no codificantes y la demostración de la existencia de mosaicismo en algunas familias3,4. Actualmente usando nuevos abordajes para el estudio genético basados en técnicas de secuenciación masiva, se llegan a caracterizar genéticamente entre el 90-98% de los pacientes con aniridia5.
En este trabajo presentamos una actualización de los aspectos epidemiológicos y genéticos, así como del algoritmo diagnóstico para abordar el estudio molecular de la aniridia. El seguimiento de las recomendaciones recogidas en este trabajo permitirá conseguir un adecuado diagnóstico a los pacientes y a sus familiares, prestar un consejo genético de calidad y mejorar el manejo clínico de esta enfermedad.
MetodologíaDurante la preparación de esta revisión, se realizó una búsqueda de las publicaciones en este tema que aparecen indexadas en Pubmed hasta el 30 de septiembre de 2020, utilizando las siguientes palabras clave: «congenital aniridia & genetics»; «PAX6 mutations», «congenital aniridia and genotype-phenotype correlation», o términos sinónimos, en los campos de título, resumen, o texto completo. Esta búsqueda no se restringió a ningún criterio temporal. Para determinar el número de nuevas mutaciones actualmente descritas en el gen PAX6, se consultaron: 1) la base de mutaciones Human PAX6 Mutation Database, cuya última actualización data del 4 de agosto de 2018 (http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6); 2) la base de mutaciones HGMD Professional (versión 2020.3), consultada el 15 de septiembre de 2020 (https://my.qiagendigitalinsights.com/bbp/view/hgmd/pro/start.php), y 3) literatura reciente que describiera nuevas variantes no registradas en estas dos bases de datos.
Aspectos epidemiológicos y clínicos de la aniridiaSe estima que la aniridia tiene una incidencia de entre 1:50.000-100.000 nacidos vivos en función de la población estudiada1. En la población española se ha descrito una incidencia mínima de 1:135.000-238.000 en el Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC) utilizando una cohorte de recién nacidos entre 1980 y 20056, cifra seguramente infraestimada dadas las dificultades de diagnóstico de la aniridia al nacimiento. Según el último informe de ORPHANET de 2020 sobre la prevalencia de enfermedades raras en Europa, la aniridia aislada tendría una incidencia estimada de 1,31/100.0007. Se desconoce la incidencia del síndrome de WAGR5, aunque se estima que el riesgo de tumor de Wilms en la aniridia es del 6%8.
A pesar de que el término «aniridia» hace referencia a la ausencia de iris, el fenotipo iridiano puede variar desde una ausencia prácticamente total a la presencia de un iris completo con pequeñas alteraciones estructurales o anomalías pupilares, pasando por una hipoplasia variable o un alargamiento iridiano mimetizando a un coloboma5,9. Los pacientes pueden presentar también anomalías del segmento anterior a lo largo de su vida como: queratopatía por disfunción limbar, ojo seco, anomalía de Peters, glaucoma juvenil asociado a alteraciones en el ángulo camerular, cataratas, ectopia lentis o subluxación del cristalino. El 90-95% de los pacientes con aniridia presentan hipoplasia foveal variable, y con menor frecuencia, hipoplasia o coloboma del nervio óptico9.
La aniridia congénita ocasionalmente se puede presentar acompañada de otras alteraciones sistémicas como una reducción olfativa, hipoacusia, alteraciones en el patrón del sueño y otras alteraciones endocrinas1, pero es necesario realizar estudios con cohortes mayores que permitan descartar un componente estocástico de los mismos. Hasta un tercio de los pacientes pueden presentar una forma sindrómica1. Los pacientes con síndrome de WAGR (MIM #194072), acrónimo del tumor de Wilms, Aniridia, anomalías Genitourinarias y Retraso del desarrollo, presentan un riesgo entre el 42-70% de desarrollar un tumor renal infantil antes de los 10 años8. Menos frecuente es la variante que asocia la obesidad, denominada síndrome de WAGRO5. Una hipoplasia severa iridiana puede aparecer en otros síndromes que cursan con disgenesia del segmento anterior como los síndromes de Axenfeld-Rieger (MIM #180500 y MIM #601499), de Peters (MIM #604229) y de Gillespie (MIM #206700), causados por genes distintos a PAX65,10. Sin embargo, la presencia de manifestaciones extraoculares típicas de estos síndromes, como anomalías dentales, cardíacas, hipoacusia, piel umbilical redundante o ataxia cerebelosa, permite realizar un diagnóstico diferencial con una aniridia sindrómica.
Etiopatogenia de la aniridiaLa aniridia está causada mayoritariamente por mutaciones en el gen Paired box-6(PAX6) que conducen a su haploinsuficiencia, es decir, a una disminución en la dosis génica que no permite mantener la función normal de este factor transcripcional. PAX6 presenta un papel fundamental en la embriogénesis ocular regulando la diferenciación espacio-temporal de los segmentos anterior y posterior del ojo11. En humanos, PAX6 se expresa de forma temprana en la vesícula óptica y posteriormente, en la copa óptica donde contribuye a la formación de la retina neural y pigmentaria11,12. También se expresa en capas ectodérmicas contribuyendo a la diferenciación de las distintas estructuras del segmento anterior, incluyendo el cristalino y el epitelio corneal12. En la etapa postnatal, su expresión queda restringida a algunas capas retinianas, cristalino, córnea, iris y cuerpo ciliar, donde PAX6 participaría en el mantenimiento de estas estructuras12. Además, PAX6 ejerce un importante papel en la morfogénesis y posterior función del sistema nervioso central, sistema olfatorio y de los islotes pancreáticos. La desregulación en la expresión de PAX6 durante el desarrollo embrionario, tanto su haploinsuficiencia como su sobreexpresión, conduce a una gran variedad de anomalías oculares en distintos modelos animales y en humanos: hipoplasia de iris, retina y nervio óptico, microftalmia, microcórnea, cataratas, etc.11,12.
PAX6, con una extensión de 22kb, se localiza en la región 11p13.3 del cromosoma 11 adyacente al gen WT1, el cual está implicado en el desarrollo del tumor de Wilms (figs. 1 y 2). PAX6 está formado por 14 exones (11 codificantes) que codifican una proteína con dos dominios de unión al ADN, un dominio pareado (PD) en el extremo N-terminal y un homeodominio (HD), además de un dominio rico en prolina-serina-treonina (PST) con función transactivadora situado en el extremo C-terminal11,12. En humanos, se han descrito 3 promotores alternativos que regulan la expresión del gen de forma espacio-temporal11,12 dando lugar a varias isoformas proteicas. Las más frecuentes son una isoforma canónica de 422 aminoácidos y una isoforma 5(a) de 436 aminoácidos por la inclusión de un exón alternativo (ex5a) que interrumpe al dominio pareado11. El balance entre ambas isoformas se ve modificado a lo largo del desarrollo regulando las diferentes funciones que PAX6 ejerce durante la embriogénesis ocular y posnatalmente13.
 Se representa la región reguladora situada aguas abajo a PAX6 (DDR, del inglés downstream regulatory region), situada en los intrones del gen vecino ELP4, con los principales elementos reguladores que han sido implicados en aniridia, SIMO y E180B (en rojo). Además, se representan los puntos de rotura de las deleciones en 11p13 detectadas previamente que ha permitido determinar la región crítica para la regulación transcripcional de PAX6. Las líneas azules y rojas discontinuas indican la región de 244Kb, definida por Ansari et al., y luego acotada a 18Kb por Plaisancié et al. B) Se representa el gen y la proteína PAX6. En la parte superior, se representa la estructura exónica con los 14 exones del gen PAX6, incluyendo regiones no codificantes (en gris) y las diferentes regiones codificantes, en donde los colores indican el dominio proteico codificado, y los promotores (P0, P1 y Pα). En la parte inferior de la imagen, se representa la estructura y los diferentes dominios funcionales de las 2 principales isoformas proteicas, la forma canónica y la isoforma 5a de 422 y 436 aminoácidos, respectivamente. HD: homeodominio; PD: dominio pareado; PST: región rica en prolina/serina/treonina.")
Representación del locus de PAX6 en la región cromosómica 11p13, gen PAX6 y sus elementos reguladores. A) Se representa la región reguladora situada aguas abajo a PAX6 (DDR, del inglés downstream regulatory region), situada en los intrones del gen vecino ELP4, con los principales elementos reguladores que han sido implicados en aniridia, SIMO y E180B (en rojo). Además, se representan los puntos de rotura de las deleciones en 11p13 detectadas previamente que ha permitido determinar la región crítica para la regulación transcripcional de PAX6. Las líneas azules y rojas discontinuas indican la región de 244Kb, definida por Ansari et al., y luego acotada a 18Kb por Plaisancié et al. B) Se representa el gen y la proteína PAX6. En la parte superior, se representa la estructura exónica con los 14 exones del gen PAX6, incluyendo regiones no codificantes (en gris) y las diferentes regiones codificantes, en donde los colores indican el dominio proteico codificado, y los promotores (P0, P1 y Pα). En la parte inferior de la imagen, se representa la estructura y los diferentes dominios funcionales de las 2 principales isoformas proteicas, la forma canónica y la isoforma 5a de 422 y 436 aminoácidos, respectivamente. HD: homeodominio; PD: dominio pareado; PST: región rica en prolina/serina/treonina.

Representación esquemática de las regiones cromosómicas implicadas en la aniridia y el síndrome de WAGR/WAGRO en el locus 11p15.1-13. Se representa la localización de los 3 principales genes implicados, WT1, PAX6 y BDNF y las regiones críticas de las deleciones 11p asociadas al síndrome de WAGR, aniridia aislada y al síndrome de WAGRO.
La activación transcripcional de PAX6 está altamente regulada a través de una compleja red de elementos reguladores que modulan su expresión génica de forma específica de tejido14,15. Hasta la fecha se han identificado una treintena de estos elementos reguladores conservados evolutivamente que están localizados en los intrones de PAX6 y en regiones intergénicas situadas antes (upstream, en localización 5’) y después (downstream, en localización 3’) del gen16 (fig. 2). La mayoría se localizan en una región 3’ situada a unas 150Kb en los intrones 7-9 del gen contiguo ELP4, la cual fue identificada gracias al estudio de los puntos de rotura en pacientes con microdeleciones en el cromosoma 11p13 en los que la región codificante del gen PAX6 se mantenía intacta17. Entre los elementos reguladores localizados en esta región destacan los elementos SIMO y E180 (fig. 2) que regulan la expresión de PAX6 durante el desarrollo de distintas estructuras oculares3,14,18. La implicación directa de SIMO en aniridia fue confirmada por Bhatia et al., 2013 al identificar una mutación puntual en este elemento que afecta al sitio de unión con PAX6 y como consecuencia, causa su inhibición transcripcional18. Estos hallazgos ponen en relieve la importancia de las regiones reguladoras en el desarrollo de aniridia.
Espectro mutacional de la aniridiaLa aniridia clásica está causada, en aproximadamente entre un 70-80% de los casos, por mutaciones codificantes en PAX6 y por microdeleciones de tamaño variable en la región 11p13-14 entre el 20-25% restante5. Hasta su última actualización en 2018, la base de datos Human PAX6 Mutation Database recoge 491 variantes distintas identificadas en este gen. Desde entonces, se han identificado unas 250 nuevas mutaciones descritas en la literatura3,4,19–30, de las cuales únicamente 190 están recogidas en la base HGMD en el momento de esta revisión. Por lo tanto, hoy en día no existe un registro de mutaciones completamente actualizado que recoja el espectro mutacional completo en PAX6.
El 85% de las mutaciones descritas se corresponden con variantes codificantes que crean un codón de parada prematuro (PTC): variantes sin sentido (nonsense) (40%), pequeñas inserciones o deleciones (30%) que alteran el marco de lectura (frameshift) y variantes de splicing, e.d. localizadas en los sitios de corte y empalme de los intrones y que afectan al procesamiento del ARN mensajero (ARNm) (15%)16. Todas estas variantes conducirían a la haploinsuficiencia de PAX6 por la degradación de ARNm portadores de PTC de forma mediada por el sistema nonsense mediated decay (NMD), un mecanismo celular de control que elimina los ARNm aberrantes. Excepto en los exones 13 y 14, se han identificado variantes PTC a lo largo de toda la secuencia codificante, destacando que el 40% están localizadas en los exones 5 y 6 que codifican el dominio proteico PD (fig. 3). Cuatro mutaciones recurrentes representan el 21% del total de los pacientes registrados en la base mutacional de PAX61,16 (fig. 3).
; en rosa, amarillo y verde, los exones que codifican los principales dominios funcionales de PAX6, pareado (PD), homeodominio (HD) y el dominio rico en prolina-serina-treonina (PST), respectivamente.")
Representación de la localización de mutaciones puntuales en la estructura génica y proteica de PAX6. Se representa el recuento de las variantes descritas en PAX6, por exones, en la base de datos PAX6 Mutation Database, hasta su última actualización en 2018, así como las variantes más representadas, incluyendo 4 mutaciones recurrentes en los exones 8, 9, 10 y 11. En gris se indican las regiones no codificantes (5’ y 3’ UTR, del inglés untranslated region); en rosa, amarillo y verde, los exones que codifican los principales dominios funcionales de PAX6, pareado (PD), homeodominio (HD) y el dominio rico en prolina-serina-treonina (PST), respectivamente.
El 15% restante de las variantes descritas no originan un PTC, incluyendo mutaciones no sinónimas que conducen a un cambio de aminoácido (missense) y variantes de pérdida del codón de terminación (stop loss). La mayoría de las mutaciones no sinónimas se localizan en residuos altamente conservados del dominio PD, y el cambio aminoacídico altera la polaridad y/o la estructura proteica16,31. Diversos trabajos centrados en el estudio in vitro de las consecuencias funcionales de variantes no sinónimas confirman que reducen la unión al ADN y la función transactivadora de PAX6 sobre genes diana, conduciendo a una pérdida de función21,32. Otras darían lugar a proteínas anómalas que actúan con un efecto dominante negativo interfiriendo con la función normal de PAX6 o incluso ganancias de función por la activación de otros genes diana diferentes a los regulados por la proteína PAX6 silvestre21,32.
En los últimos años se ha descrito la implicación de variantes en regiones reguladoras y no codificantes del gen PAX6 en la aniridia3,20,28. La mayoría se localizan en sitios de splicing de los exones 2 y 33,16 (fig. 3), que forman parte de la región no codificante del gen situada a 5’ del codón de inicio de la traducción (5’UTR, del inglés untranslated region). Además, se han identificado variantes en regiones intrónicas profundas (deep-intronic) y variantes sinónimas en las que se predice la creación de splicing alternativos mediante la creación de sitios de splicing no canónicos y/o la inclusión de nuevos seudoexones3,20,33. El estudio in vitro de estas variantes no codificantes mediante la estrategia de «minigenes», que utiliza vectores artificiales de splicing que incluyen uno o varios exones de un gen, separados por secuencias intrónicas, así como sitios funcionales de splicing necesarios para su expresión, está siendo clave para confirmar su patogenicidad mediada por desregulación de los patrones normales de splicing de PAX63,28,33. Además, se han identificado variantes génicas en los elementos reguladores que podrían alterar la expresión transcripcional de PAX63,18.
Por otro lado, hasta el 25% de los casos de aniridia están causados por variantes estructurales, e.d. inserciones, deleciones o duplicaciones de segmentos de ADN, tal como reflejan estudios realizados en distintas cohortes5,16,33–35. Las más frecuentes son microdeleciones de tamaño variable en la región 11p13-14 que pueden afectar al gen PAX6 o únicamente a sus elementos reguladores situados a 3’, así además pueden abarcar un número variable de genes contiguos a PAX61,16,36. Estas deleciones de genes contiguos incluyen al menos a WT1 en pacientes con síndrome de WAGR y, además, al gen BDFN asociado a obesidad en el síndrome de WAGRO, localizados a una distancia de unos 700Kb y 4,2Mb de PAX6, respectivamente1,5,37 (fig. 2). La inclusión en estas deleciones de los genes PRGG4 y LMO2 podría modular la aparición de discapacidad intelectual o aumentar el riesgo a desarrollar tumor de Wilms24,38. Sin embargo, se desconoce la participación en el fenotipo de los más de 40 genes que podrían estar incluidos en estas deleciones. Con menor frecuencia pueden existir otras variantes estructurales como translocaciones, inversiones o duplicaciones del cromosoma 11p13 involucrando PAX6 o sus elementos reguladores (figs. 1 y 2) y que se han asociado a anomalías oculares y sistémicas39,40.
Correlaciones genotipo-fenotipoLas variantes en PAX6 conducen a distintas presentaciones clínicas en función de la naturaleza de la mutación y de su localización en el gen16. Existe una gran variabilidad fenotípica inter- e intrafamiliar en los pacientes portadores de PTC, por lo que no existen claras correlaciones genotipo-fenotipo9,16,41. Sin embargo, se suele aceptar que las mutaciones que conducen a la ganancia o pérdida de un PTC se asocian a un fenotipo de aniridia clásica con hipoplasia iridiana y macular (tabla 1). En un estudio reciente en una cohorte de 46 pacientes, se apunta a posibles correlaciones entre el tipo de variante y la queratopatía asociada a la aniridia, con una mayor severidad y una presentación más temprana en portadores de deleciones completas del gen y, por el contrario, formas más leves y no progresivas asociada a mutaciones no sinónimas23,42. Por el contrario, no se observó esta asociación en una cohorte de 126 pacientes estudiada por Vasilyeva et al., aunque sí describen que los portadores de deleciones que afectan a los elementos reguladores de PAX6 presentan fenotipos más leves de hipoplasia foveal y queratopatía22. Por lo tanto, sería necesario estudiar grandes cohortes de pacientes con evaluaciones clínicas más homogéneas que permitan confirmar o descartar estas observaciones.
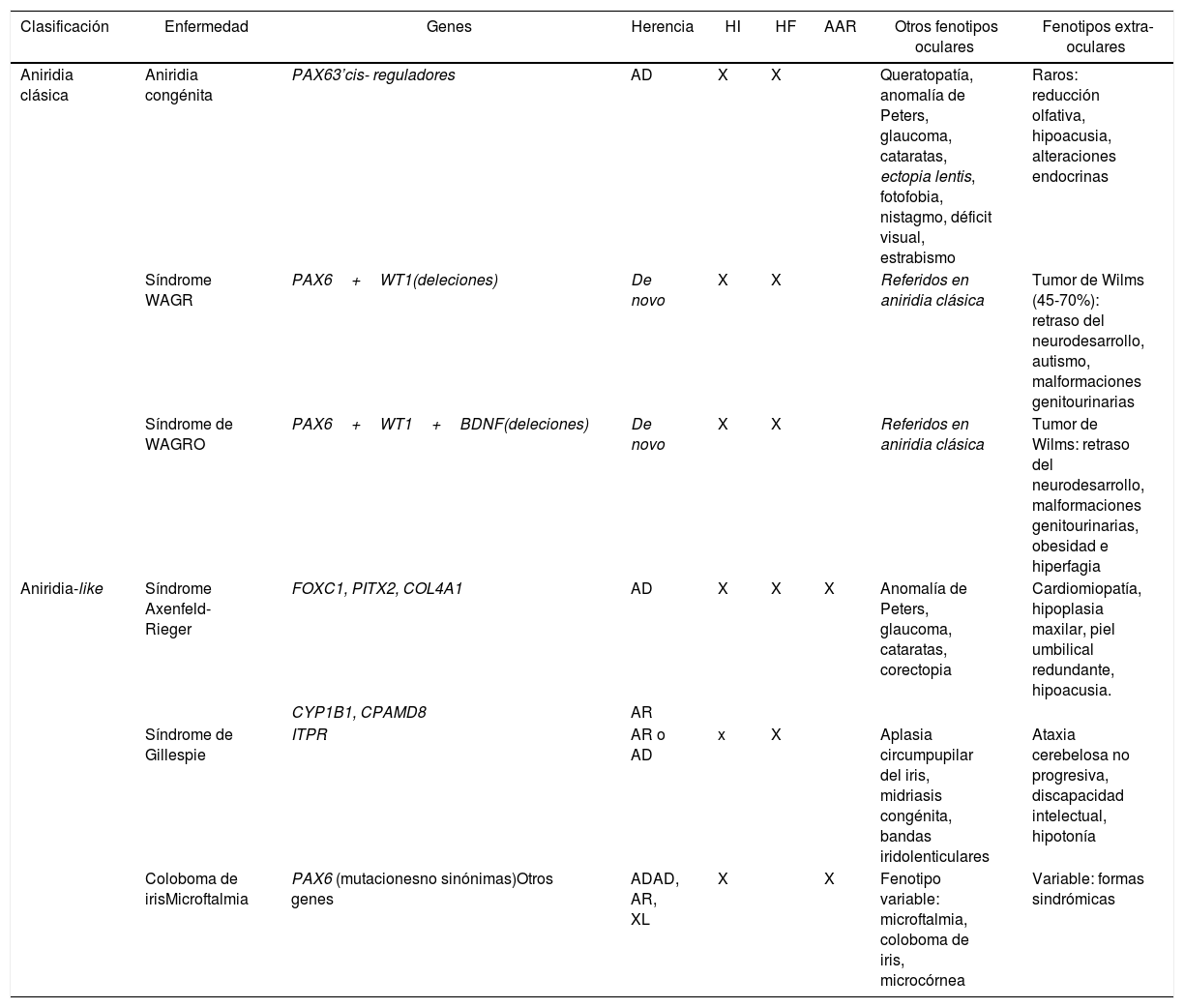
Clasificación fenotipo-genotipo de formas de aniridia y diagnóstico diferencial con otras formas oculares
| Clasificación | Enfermedad | Genes | Herencia | HI | HF | AAR | Otros fenotipos oculares | Fenotipos extra-oculares |
|---|---|---|---|---|---|---|---|---|
| Aniridia clásica | Aniridia congénita | PAX63’cis- reguladores | AD | X | X | Queratopatía, anomalía de Peters, glaucoma, cataratas, ectopia lentis, fotofobia, nistagmo, déficit visual, estrabismo | Raros: reducción olfativa, hipoacusia, alteraciones endocrinas | |
| Síndrome WAGR | PAX6+WT1(deleciones) | De novo | X | X | Referidos en aniridia clásica | Tumor de Wilms (45-70%): retraso del neurodesarrollo, autismo, malformaciones genitourinarias | ||
| Síndrome de WAGRO | PAX6+WT1+BDNF(deleciones) | De novo | X | X | Referidos en aniridia clásica | Tumor de Wilms: retraso del neurodesarrollo, malformaciones genitourinarias, obesidad e hiperfagia | ||
| Aniridia-like | Síndrome Axenfeld-Rieger | FOXC1, PITX2, COL4A1 | AD | X | X | X | Anomalía de Peters, glaucoma, cataratas, corectopia | Cardiomiopatía, hipoplasia maxilar, piel umbilical redundante, hipoacusia. |
| CYP1B1, CPAMD8 | AR | |||||||
| Síndrome de Gillespie | ITPR | AR o AD | x | X | Aplasia circumpupilar del iris, midriasis congénita, bandas iridolenticulares | Ataxia cerebelosa no progresiva, discapacidad intelectual, hipotonía | ||
| Coloboma de irisMicroftalmia | PAX6 (mutacionesno sinónimas)Otros genes | ADAD, AR, XL | X | X | Fenotipo variable: microftalmia, coloboma de iris, microcórnea | Variable: formas sindrómicas |
AAR: anomalía de Axelfeld-Rieger; AD: autosómico dominante; AR: autosómico recesivo; HF: hipoplasia foveal; HI: hipoplasia de iris; XL: Ligado al cromosoma X (X-linked).
Por otro lado, es difícil predecir el fenotipo asociado a las mutaciones no sinónimas16,41. Estas se identifican en pacientes tanto con formas leves con manifestación parcial del espectro fenotípico (hipoplasia aisladas de iris, fóvea o nervio óptico)9,27,31,43, como con formas severas de aniridia clásica22 (tabla 1). Además, pueden asociarse a fenotipos aislados o sindrómicos de microftalmia colobomatosa4,21,31,43. Un reciente estudio describe que presentaciones severas de microftalmia sindrómica asociadas a PAX6 aparecen en portadores de mutaciones no sinónimas que alteran la interacción del dominio PD de PAX6 con el factor de transcripción SOX2, cuyas mutaciones conducen a un fenotipo muy similar21.
Diagnóstico genético en la aniridiaLos estudios genéticos en aniridia permiten confirmar la sospecha clínica, realizar diagnóstico diferencial en casos con clínica dudosa, conocer el riesgo de desarrollar tumor de Wilms en casos esporádicos de recién nacidos o en la primera infancia con diagnóstico de aniridia, proporcionar asesoramiento genético, establecer la evolución y pronóstico en la medida de lo posible, en función del tipo de mutación y abrir la posibilidad de realizar diagnóstico genético prenatal y/o preimplantacional1,2,44. Las recomendaciones y consideraciones previas a la realización de un estudio genético en aniridia y síndrome WAGR se pueden consultar en 2 guías para el estudio genético de la aniridia y del síndrome WAGR publicadas anteriormente2,45. El diagnóstico genético de la aniridia debe incluir un estudio dirigido del gen PAX6 para cribar los distintos tipos de variantes en función de la herencia y edad del paciente2,44 (fig. 4).
. El objetivo principal es descartar mutaciones y/o deleciones que causen haploinsuficiencia de PAX6 en casos con sospecha de aniridia y con fenotipo clásico, además de un diagnóstico diferencial con otros genes, principalmente de disgenesia de segmento anterior, en casos de aniridia con presentaciones atípicas. Se prioriza el estudio de variaciones en el número de copia en el cromosoma 11p en casos esporádicos menores a 10 años y/o con presentación de síntomas extraoculares, característicos del síndrome de WAGR (tumor de Wilms, retraso del neurodesarrollo, anomalías genitourinarias), mediante distintas técnicas moleculares, o en su defecto citogenéticas. El cribado de mutaciones en PAX6 se priorizará en casos familiares o esporádicos mayores de 10 años por su mayor rendimiento. En este algoritmo incorporamos el uso de técnicas de NGS (paneles dirigidos a PAX6 y otros genes asociados a malformaciones oculares, exomas clínicos, exomas completos o genomas) para realizar ambos análisis, siempre teniendo en cuenta las limitaciones de sensibilidad de cada técnica en la valoración del test genético.")
Diagrama de flujo del diagnóstico genético de la aniridia propuesto de diagnóstico genético en aniridia. Se propone un algoritmo diagnóstico en función de la sospecha clínica y características del paciente (edad y antecedentes familiares). El objetivo principal es descartar mutaciones y/o deleciones que causen haploinsuficiencia de PAX6 en casos con sospecha de aniridia y con fenotipo clásico, además de un diagnóstico diferencial con otros genes, principalmente de disgenesia de segmento anterior, en casos de aniridia con presentaciones atípicas. Se prioriza el estudio de variaciones en el número de copia en el cromosoma 11p en casos esporádicos menores a 10 años y/o con presentación de síntomas extraoculares, característicos del síndrome de WAGR (tumor de Wilms, retraso del neurodesarrollo, anomalías genitourinarias), mediante distintas técnicas moleculares, o en su defecto citogenéticas. El cribado de mutaciones en PAX6 se priorizará en casos familiares o esporádicos mayores de 10 años por su mayor rendimiento. En este algoritmo incorporamos el uso de técnicas de NGS (paneles dirigidos a PAX6 y otros genes asociados a malformaciones oculares, exomas clínicos, exomas completos o genomas) para realizar ambos análisis, siempre teniendo en cuenta las limitaciones de sensibilidad de cada técnica en la valoración del test genético.
Ante casos de aniridia esporádica en menores de 9 años sin antecedentes de enfermedad ocular en los progenitores, es prioritario realizar un estudio genético que permita descartar microdeleciones en 11p13 que impliquen al gen WT1, con el fin de conocer la predisposición a desarrollar tumor de Wilms2,44,45 (figs. 2 y 4). Para ello, existen distintos abordajes moleculares y citogenéticos, destacando la técnica de Multiplex ligation-dependent probe amplification (MLPA) por su rapidez y bajo coste. Existen diseños comerciales de MLPA que analizan microdeleciones y otras variantes estructurales en PAX6, sus regiones reguladoras, WT1 y otros genes localizados en 11p13-141,2. Sin embargo, esta aproximación no permite acotar ni el tamaño ni los puntos de rotura exactos de las deleciones. Para ello, recomendamos los arrays de hibridación genómica comparada (aCGH), tanto comerciales como los diseñados específicamente para analizar el locus WAGR2,34, enriquecidos con un mayor número de sondas en esta región lo que permite acotar el tamaño con mayor precisión34. En general, estas técnicas moleculares permiten detectar microdeleciones entre el 15-25% de los pacientes, principalmente en casos esporádicos5,22,33. No obstante, no pueden detectar inversiones, translocaciones o reordenamientos equilibrados, por lo que su uso no invalida la utilidad de las técnicas citogenéticas clásicas, como el cariotipo de alta resolución y la FISH en pacientes no caracterizados genéticamente tras el uso de técnicas moleculares1,2,39.
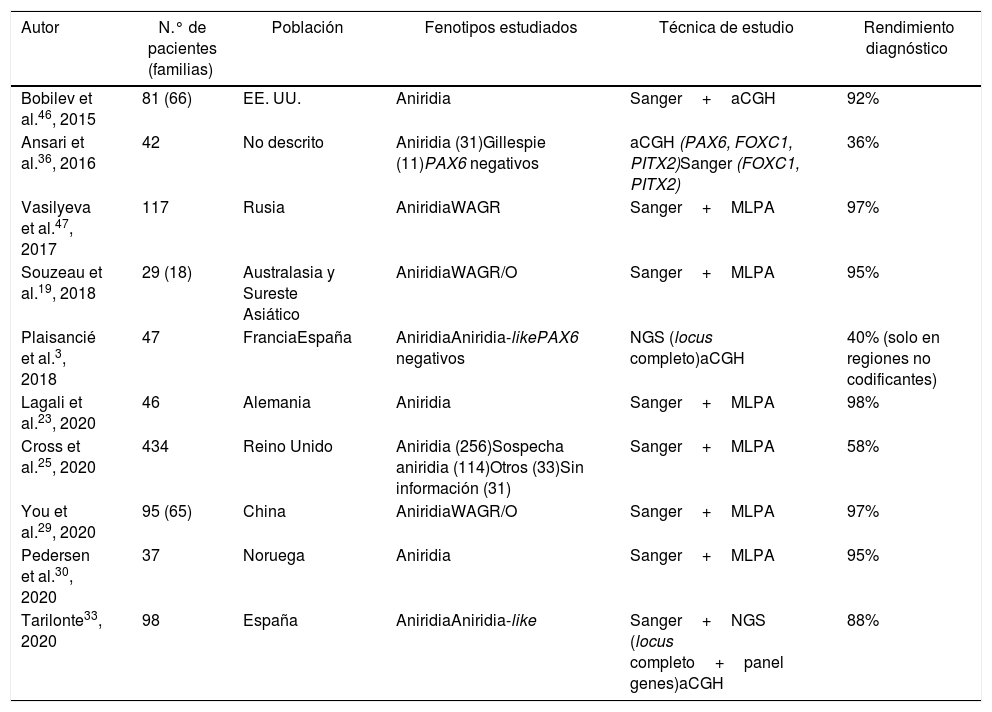
Diagnóstico molecular de variantes puntuales en la secuencia de PAX6En casos familiares y casos esporádicos no portadores de microdeleciones en 11p13-14, debe priorizarse el estudio de mutaciones puntuales en la secuencia codificante de PAX6, e.d. los exones 4-142,44 (fig. 1). La técnica más utilizada hasta la fecha es la secuenciación Sanger con un rendimiento de detección entre el 50-90% en función de la cohorte en estudio y las regiones de PAX6 analizadas (tabla 2)1,22,25,30,33,44,46,47.
Cohortes de pacientes con aniridia en distintas poblaciones
| Autor | N.° de pacientes (familias) | Población | Fenotipos estudiados | Técnica de estudio | Rendimiento diagnóstico |
|---|---|---|---|---|---|
| Bobilev et al.46, 2015 | 81 (66) | EE. UU. | Aniridia | Sanger+aCGH | 92% |
| Ansari et al.36, 2016 | 42 | No descrito | Aniridia (31)Gillespie (11)PAX6 negativos | aCGH (PAX6, FOXC1, PITX2)Sanger (FOXC1, PITX2) | 36% |
| Vasilyeva et al.47, 2017 | 117 | Rusia | AniridiaWAGR | Sanger+MLPA | 97% |
| Souzeau et al.19, 2018 | 29 (18) | Australasia y Sureste Asiático | AniridiaWAGR/O | Sanger+MLPA | 95% |
| Plaisancié et al.3, 2018 | 47 | FranciaEspaña | AniridiaAniridia-likePAX6 negativos | NGS (locus completo)aCGH | 40% (solo en regiones no codificantes) |
| Lagali et al.23, 2020 | 46 | Alemania | Aniridia | Sanger+MLPA | 98% |
| Cross et al.25, 2020 | 434 | Reino Unido | Aniridia (256)Sospecha aniridia (114)Otros (33)Sin información (31) | Sanger+MLPA | 58% |
| You et al.29, 2020 | 95 (65) | China | AniridiaWAGR/O | Sanger+MLPA | 97% |
| Pedersen et al.30, 2020 | 37 | Noruega | Aniridia | Sanger+MLPA | 95% |
| Tarilonte33, 2020 | 98 | España | AniridiaAniridia-like | Sanger+NGS (locus completo+panel genes)aCGH | 88% |
La reciente implementación de técnicas de secuenciación de nueva generación (Next Generation Sequecing [NGS]) permite analizar una gran cantidad de regiones de ADN en paralelo y con ello, realizar estudios con mayor coste-efectividad y rapidez5. Hoy en día, estas técnicas de NGS representan en nuestro laboratorio el primer abordaje para el estudio de la aniridia y enfermedades relacionadas3,4,33. El uso de diseños personalizados («panel de aniridia») permite secuenciar el locus completo de PAX6, incluyendo exones, UTR, intrones y regiones reguladoras3, con lo que se alcanza un rendimiento diagnóstico entre el 85-95%3,5,33, mayor que el obtenido utilizando aproximaciones comerciales basadas en la secuenciación de exoma clínico. Por otro lado, la disminución del coste de la secuenciación NGS y la mejora de los análisis bioinformáticos posibilita que en algunos laboratorios, se esté secuenciando en pacientes con aniridia el exoma completo (Whole Exome Sequencing [WES])26,28 e incluso el genoma completo (Whole Genome Sequencing [WGS])48. Esta última aproximación cubre regiones no codificantes y tiene una mayor sensibilidad para detectar variaciones estructurales. En función del diseño y el análisis bioinformático realizado, estas aproximaciones de NGS podrían detectar de manera simultánea variantes puntuales y estructurales3, lo que permitirá en un futuro cercano abordar el estudio de la aniridia con un único test genético.
Diagnóstico diferencialA pesar del carácter congénito de la aniridia, la obtención de un diagnóstico clínico precoz puede verse dificultado por la elevada variabilidad en sus manifestaciones clínicas tempranas y su alto grado de solapamiento con otras enfermedades oculares (tabla 1). En el caso de bebés con fotofobia, nistagmo y sospecha de baja visión, en los que resulta difícil realizar una valoración oftalmológica, los estudios genéticos permiten realizar un diagnóstico diferencial con otras enfermedades con clínica solapante: coloboma iridiano, albinismo ocular, hipoplasia foveal, cataratas congénitas, esclerocórnea congénita y en especial, con disgenesias del segmento anterior que pueden asociar hipoplasias iridianas severas5,10 (fig. 4 y tabla 1).
Algunos pacientes con fenotipo leve de aniridia o aniridia-like no portadores de mutaciones en el gen PAX6 pueden presentar variantes en genes de iridodisgenesias tanto asociados a formas dominantes (FOXC1, PITX2 y COL4A1)10,36, como a recesivas (CYP1B1 y CPAMD8)10,49 (tabla 1). En pacientes con aplasia circumpupilar del iris, se deben descartar mutaciones dominantes y recesivas en el gen ITPR1 responsable del síndrome de Gillespie, caracterizado por ataxia cerebelar no progresiva y discapacidad intelectual5 (tabla 1). El uso de paneles génicos que contienen estos genes y otros implicados en enfermedades solapantes de malformaciones oculares nos ha permitido identificar nuevas mutaciones y/o variantes estructurales y describir una nueva asociación fenotípica con el gen de cataratas CRYAA33,49,50. Por todo ello, recomendamos realizar diagnóstico diferencial utilizando paneles extendidos de aniridia en casos con fenotipos atípicos y en especial, en aquellos casos sin mutaciones en PAX633 (fig. 4).
Herencia y asesoramiento genético en la aniridiaLa aniridia clásica se transmite con un patrón de herencia autosómica dominante, e.d. solo un alelo debe estar alterado para que se ponga de manifiesto el problema ocular. Suele existir segregación familiar con varias generaciones de afectos en dos tercios de los casos. El riesgo de transmisión de un afectado a su descendencia independiente del sexo será del 50% (fig. 4).
Un tercio de los casos son esporádicos debido a la aparición de una variante de novo que suele estar ausente en los progenitores no afectados, pero es altamente recomendable confirmarlo mediante un estudio de segregación. A pesar de que el riesgo de repetición es muy bajo tras el nacimiento de un caso esporádico, no se puede descartar completamente la posibilidad de mosaicismo germinal en alguno de los progenitores para una mutación aparentemente de novo. Recientemente, hemos descrito por primera vez la presencia de mosaicismo parental en varias familias con mutaciones en PAX64,27,33. Ante la sospecha de un mosaicismo somático o gonosomal en una familia, el análisis cuantitativo de la mutación utilizando técnicas de PCR digital en tejidos de distinto origen embrionario puede ser útil para calcular el riesgo de recurrencia4. Por otra parte, ante un caso de novo, de cara a las futuras gestaciones se establece un riesgo de recurrencia pequeño pero real, por lo que se recomienda seguimiento prenatal, de ser factible mediante diagnóstico prenatal no invasivo (DPNI).
En el caso de portadores de microdeleciones 11p13-14, estas suelen estar asociadas a eventos de novo en el cromosoma paterno35, pero sigue siendo recomendable realizar un cariotipo que descarte reordenamientos equilibrados en 11p13.
Diagnóstico prenatal y preimplantacionalLa identificación de la causa genética en un paciente con aniridia abre la posibilidad de realizar estudios prenatales y preimplantacionales en la familia para evitar la transmisión de una mutación familiar o en el caso de mutaciones aparentemente de novo, de un eventual mosaico germinal. Para ello, es necesario realizar un adecuado consejo genético donde se valoren las opciones más adecuadas en función del tipo de mutación, situación reproductiva, edad de los progenitores y de la factibilidad técnica para detectar la variante2. El diagnóstico prenatal se suele realizar con aproximaciones invasivas a partir de biopsia de vellosidad corial o amniocentesis44. Actualmente, existe la posibilidad de realizar DPNI mediante el estudio del ADN fetal en sangre materna, indicado para mutaciones de novo o heredadas por vía paterna2.
Perspectivas futurasA pesar de los grandes avances en el conocimiento de los mecanismos etiopatogénicos de la aniridia, existen aún muchas cuestiones por resolver en relación con las correlaciones genotipo-fenotipo y con la gran variabilidad fenotípica causada por mutaciones similares en el gen PAX6. Dada la complejidad en la regulación transcripcional de PAX6 y su participación en múltiples redes morfogénicas espacio-temporal durante el desarrollo ocular, se desconoce la influencia de los elementos reguladores o de genes moduladores de la expresión génica que podrían contribuir en parte a la expresividad variable en la aniridia. El mosaicismo somático podría explicar también algunos casos de variabilidad fenotípica intrafamiliar4. A medida que mejore la sensibilidad técnica para detectar alelos de baja frecuencia, esperamos que el estudio del mosaicismo pueda integrarse en el futuro en el diagnóstico genético de la aniridia y así, ofrecer un mejor asesoramiento genético en casos con mutaciones de novo. Otro aspecto para tener en cuenta es la caracterización de los casos sin mutaciones en PAX6. El uso de nuevas tecnologías de WGS y el de otros abordajes permitirá estudiar las regiones reguladoras del gen PAX6 y de otros genes, además de desenmascarar nuevos casos de mosaicos somáticos que hayan podido pasar desapercibidos debido a las limitaciones actuales en las técnicas de secuenciación.
FinanciaciónEste trabajo ha sido financiado con fondos procedentes del Instituto de Salud Carlos III, parcialmente financiados por Fondos Europeos para el desarrollo Regional (PI17/01164, PI20/00851, PI17/1659, Miguel Servet CPII17_00006, FI18_00123, CIBERER CB06/07/0036), por la Comunidad de Madrid (RareGenomics B2017/BMD3721, EJD-2018-PRE_BMD-9453), la Federación Española de Enfermedades Raras (FEDER) y la ONCE (P12-18, P20).
Conflicto de interesesLos autores declaran que no existe ningún conflicto de intereses.
Queremos agradecer a los pacientes con aniridia su colaboración en los proyectos de investigación, así como a los profesionales sanitarios, genetistas, pediatras, oftalmólogos y técnicos de laboratorio que han participado en el diagnóstico clínico y molecular de estos pacientes, así como el facilitarnos la obtención de muestras y datos clínicos. Agradecemos el apoyo prestado por la Asociación Española de Aniridia (AEA), a lo largo de estos años, así como a la asociación Aniridia Europa.
