






Introducción
La publicación de casos aislados o de pequeñas series de casos de reacciones adversas a medicamentos (RAM) es un instrumento utilizado tradicionalmente por los profesionales sanitarios para intercambiar conocimientos. La publicación en la revista The Lancet de una carta al director1 con la observación de un aumento de frecuencia de casos de focomelia en niños cuyas madres habían tomado talidomida durante el embarazo marcó un hito en la historia de la regulación de los medicamentos y de la farmacovigilancia. Este grave problema de seguridad fue el origen de la creación de un programa internacional permanente de notificación sistematizada de sospechas de reacciones adversas a medicamentos. Este programa debía resolver dos de los problemas de la publicación de casos; uno, la decisión del editor de publicarlo o no y otro, el retraso de la publicación desde el diagnóstico. El Sistema Español de Farmacovigilancia (SEFV) participa en este programa para la identificación de problemas de seguridad no detectados antes de la comercialización de los medicamentos.
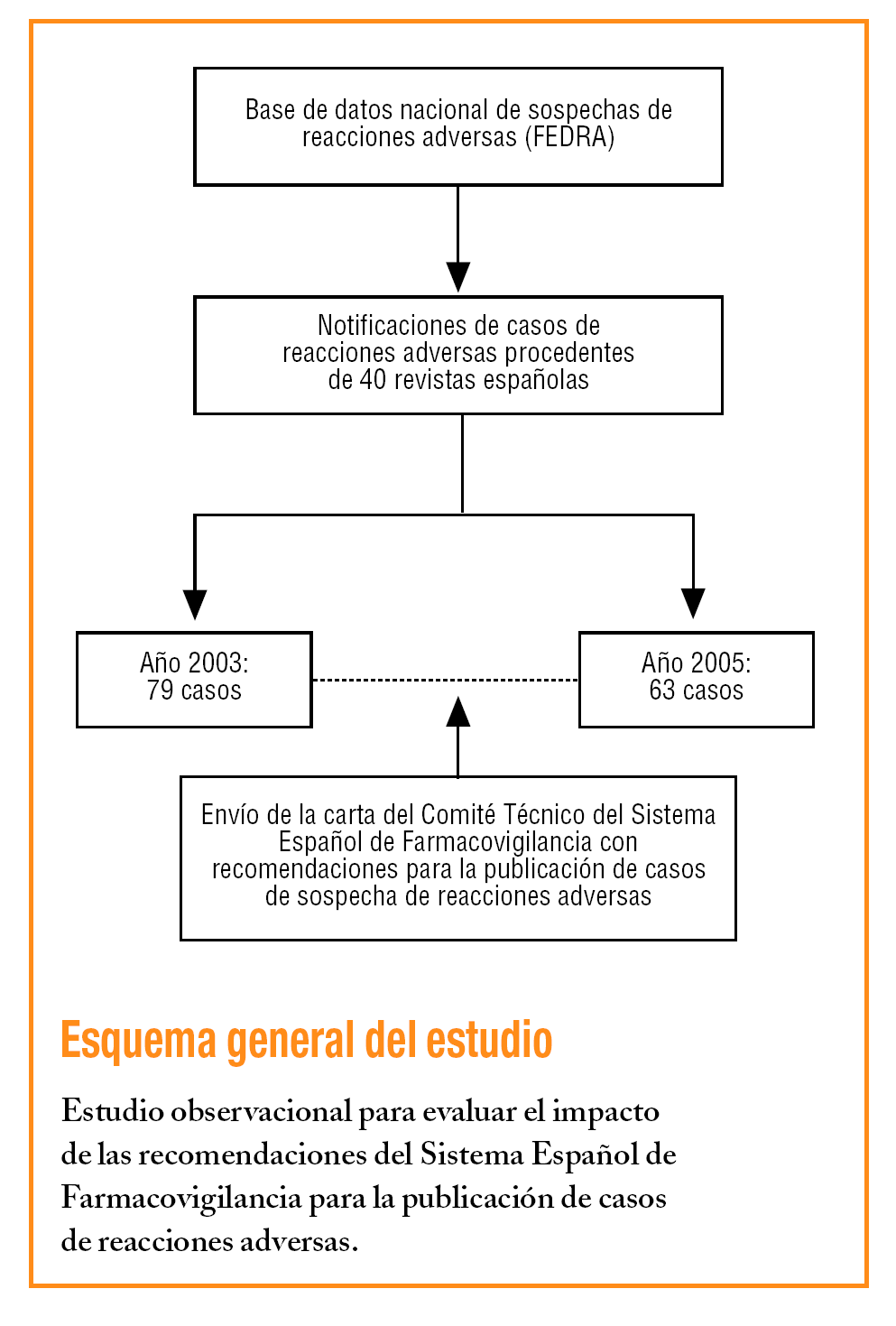
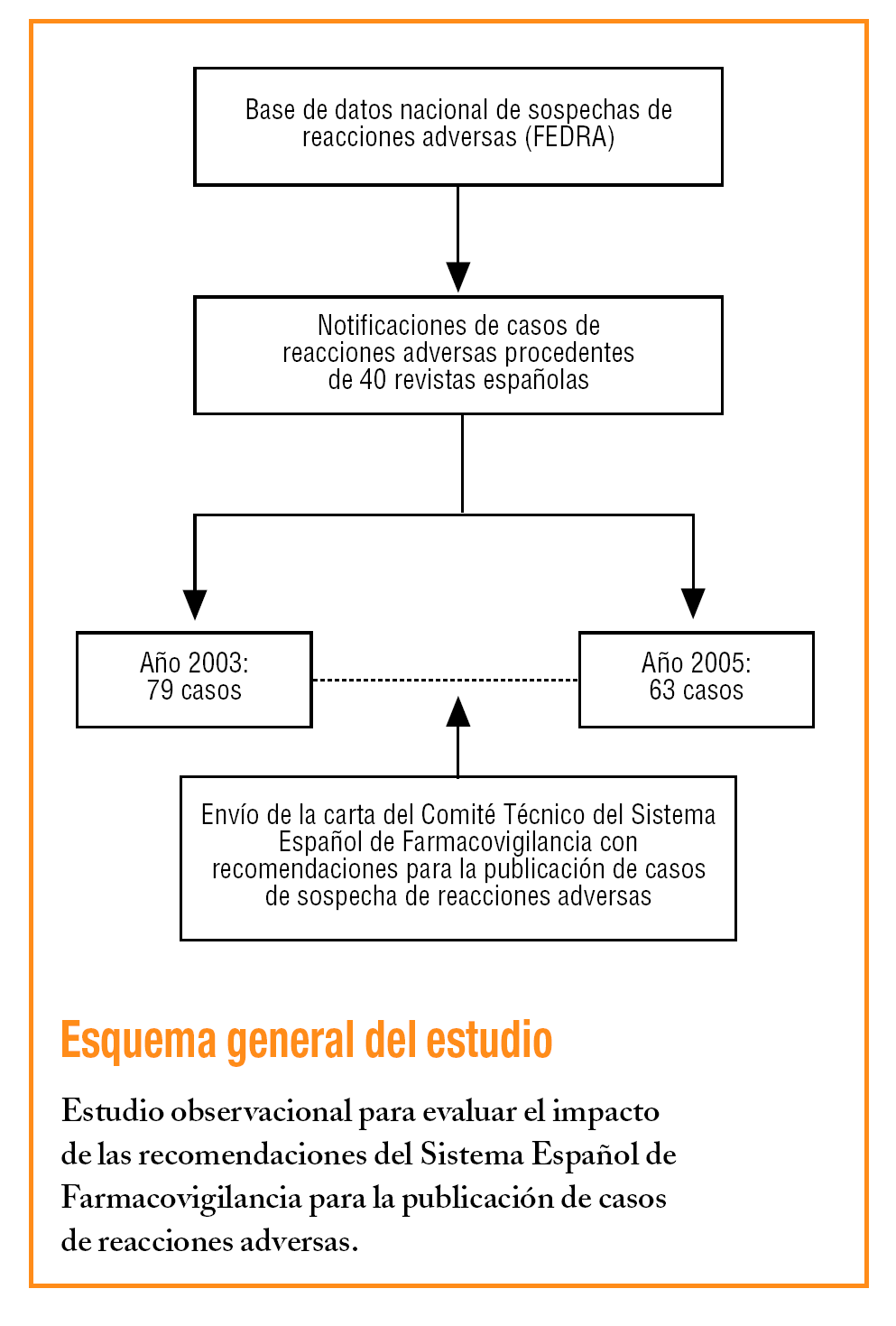
Cada uno de los 17 centros autonómicos de farmacovigilancia que forman el SEFV evalúa las notificaciones enviadas por los profesionales sanitarios. Además, revisan los casos de sospechas de reacciones adversas de 40 revistas biomédicas españolas y, si no han sido previamente notificados, son evaluados e introducidos en la base nacional de sospechas de reacciones adversas a medicamentos (FEDRA). Estos casos pueden contribuir a la generación de una señal de alerta, es decir, a lo que la Organización Mundial de la Salud (OMS) define como la información comunicada de una posible relación causal entre un acontecimiento adverso y un fármaco, cuando previamente esta relación era desconocida o estaba documentada de forma incompleta2. Habitualmente se requiere más de una notificación para generar una señal, dependiendo de la gravedad del acontecimiento y de la calidad de la información2. Es importante que de la publicación se pueda extraer la información necesaria para evaluar el caso e incorporarlo de forma estandarizada en la base de datos. El Comité Técnico del Sistema Español de Farmacovigilancia (CTSEFV), órgano que unifica los criterios de funcionamiento y evalúa las señales generadas por el Sistema Español de Farmacovigilancia, fue informado de que la calidad de la información necesaria para la evaluación de cada caso era inferior en las sospechas de reacciones adversas recogidas en FEDRA que proceden de la revisión de la literatura que las notificadas directamente por el profesional sanitario mediante la tarjeta amarilla, el 39% contiene la información mínima frente al 67%, respectivamente3.
Por este motivo, en noviembre de 2003 el CTSEFV revisó las recomendaciones ya recogidas en las Buenas prácticas de farmacovigilancia del Sistema Español de Farmacovigilancia, publicadas en el año 20004. En mayo de 2004, desde el CTSEFV, se envió a los editores de esas 40 revistas una carta con las recomendaciones para los autores (tabla 1) y revisores sobre cómo presentar los casos de sospechas de reacciones adversas.

El presente trabajo tiene como objetivo evaluar la repercusión de las recomendaciones del CTSEFV en la calidad de la descripción de los casos de sospechas de reacciones adversas a medicamentos publicados en revistas biomédicas españolas.
Métodos
Se seleccionaron las sospechas de reacciones adversas de las revistas españolas que revisan los centros de farmacovigilancia, incorporadas en FEDRA hasta el 21 de diciembre de 2005. Se comparó la calidad de la información de los casos publicados el año previo (2003) y el posterior (2005) al año de envío de las recomendaciones.
En cada notificación se revisó la información incluida en los apartados: sexo, edad, dosis, motivo de prescripción, duración del tratamiento, duración de la reacción adversa, secuencia temporal, efecto de la retirada y causas alternativas. Se definieron los parámetros para considerar inadecuada la información de los apartados anteriores. La duración del tratamiento se consideró inadecuada cuando en las fechas de inicio o final sólo constaba el año, o la fecha final era desconocida. La duración de la reacción se consideró inadecuada cuando en las fechas de inicio y final sólo aparecía el año o cuando el final de la reacción era desconocido. El efecto de la retirada se consideró inadecuado cuando en la evaluación que realizan los técnicos del SEFV se le asignó el código correspondiente a «no hay información respecto a la retirada del fármaco o de los efectos de la retirada». Las causas alternativas al fármaco se consideraron inadecuadas cuando en la evaluación de los técnicos se le asignó el código correspondiente a «no hay información suficiente en la notificación para poder evaluar la relación causal, aunque ésta se puede sospechar».
Se creó un indicador global3 consistente en el cumplimiento simultáneo de las variables sexo, edad, dosis, motivo de prescripción, secuencia temporal y efecto de la retirada para los años 2003 y 2005. La comparación entre los años 2003 y 2005 se ha realizado con la prueba de la c2.
Resultados
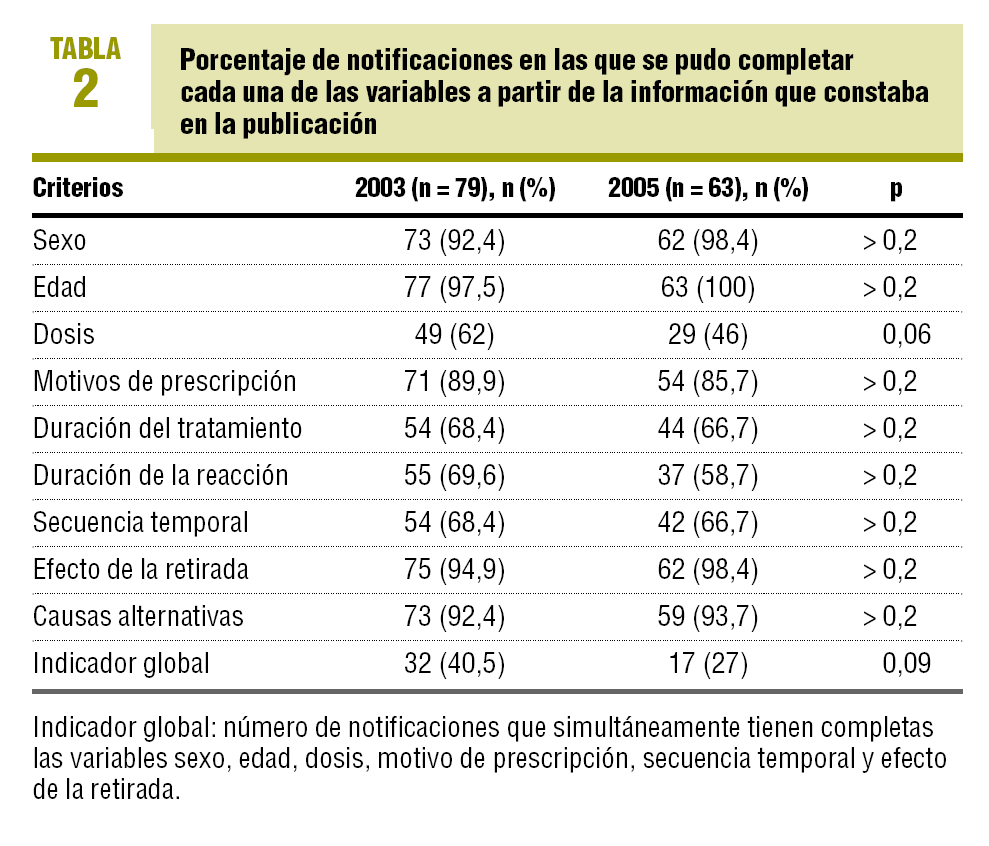
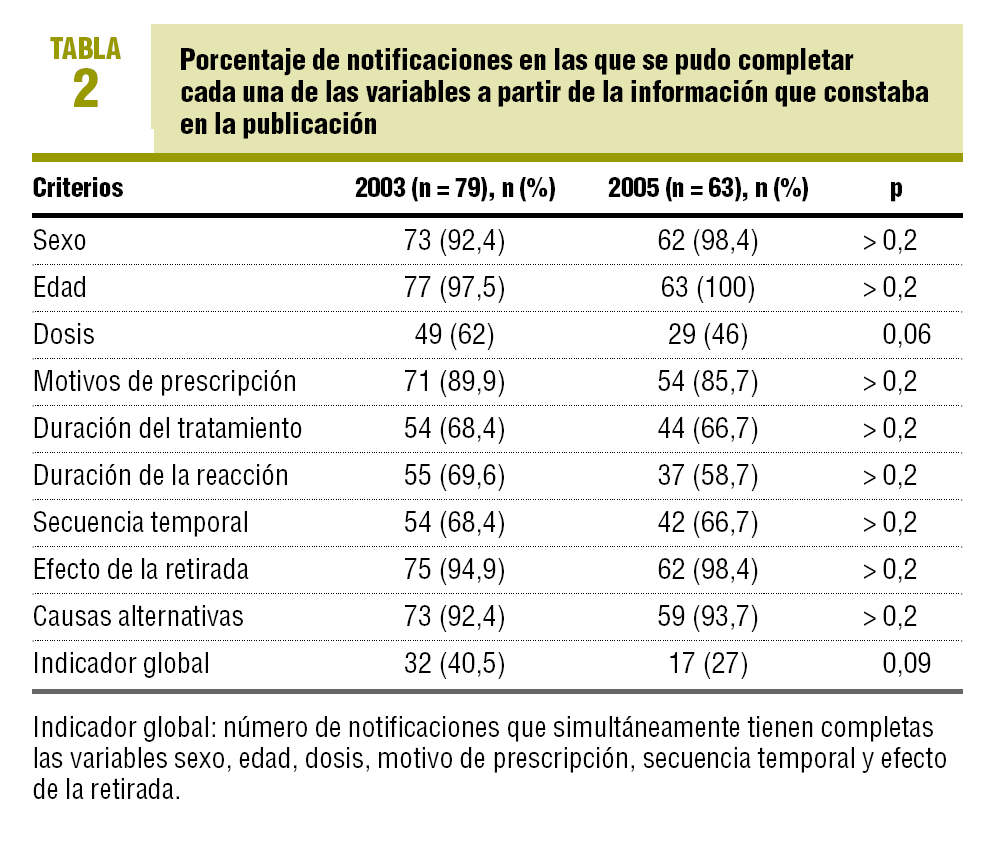
Hasta el 21 de diciembre de 2005 en FEDRA estaban recogidos 79 casos de sospechas de reacciones adversas de casos publicados en el 2003 y 63 casos publicados en el año 2005. En la tabla 2 están recogidos el porcentaje de casos en los que se cumple cada variable, destaca el apartado dosis con el 46% de los casos en 2005, frente al 62% en 2003, y la duración de la RAM en el 58,8% en 2005, frente al 69,6% en 2003. En la comparación de cada una de las variables analizadas entre los años 2005 y 2003 no se detecta que las diferencias sean estadísticamente significativas. El indicador global para el cumplimiento de las seis variables fue de un 27% en 2005, frente a un 40,5% en 2003, esta diferencia tampoco es estadísticamente significativa.
Discusión
Únicamente el 27% de las RAM recogidas en FEDRA procedentes de casos publicados en el año 2005 contienen información completa en todos los apartados del indicador global. Aunque no hay diferencias estadísticamente significativas respecto a las publicaciones de 2003, probablemente por el bajo número de casos, la tendencia es hacia un empeoramiento de la calidad de la información. Tanto en los años analizados en este trabajo como en el análisis previo, de los años 2000 y 20013, los apartados con un mayor porcentaje de falta de información son las fechas de tratamiento, las fechas de reacción y, como consecuencia, la secuencia temporal. Por lo tanto, es difícil establecer si la relación temporal entre la administración de los medicamentos y la aparición del acontecimiento adverso es coherente con el mecanismo de acción del fármaco o con el proceso fisiopatológico de la reacción adversa. Otro apartado que no se recoge adecuadamente es la dosis administrada al paciente, lo que impide evaluar si la posible señal de alerta depende de la dosis. La falta de información en los apartados dosis y duración del tratamiento, observada en esta revisión, concuerda con los datos publicados por Gil López-Oliva et al5 correspondientes al período 1992-1994.
Sin embargo, nuestros datos contrastan con una revisión publicada por Sempere et al6. Los resultados de calidad que obtienen son claramente superiores a los del presente trabajo para las mismas variables. Estas discrepancias se podrían explicar por los diferentes criterios utilizados para considerar adecuada la información o por otras diferencias metodológicas. Sempere et al analizan los casos a partir de las publicaciones y sólo revisan 4 revistas, en el presente trabajo se analiza la información que se ha podido trasladar de forma sistematizada a una base de datos procedente de 40 revistas españolas revisadas por los centros de farmacovigilancia.
El grado de cumplimiento de los criterios requeridos para evaluar las notificaciones determina el número de casos necesarios para generar una señal de alerta7. Cuanto mayor y más completa es la información aportada por cada caso de sospecha de RAM, menor es el número de casos necesarios para generar una señal de alerta.
La preocupación por la necesidad de estandarizar la información que debe constar en las publicaciones de sospechas de reacciones adversas sigue siendo un tema de actualidad, por lo que recientemente un grupo de trabajo auspiciado por la Sociedad Internacional de Farmacoepidemiología (ISPE) y la Sociedad Internacional de Farmacovigilancia (ISoP) ha elaborado y difundido unas directrices en este sentido8.
Por otra parte, la publicación de un caso de sospecha de reacción adversa es perfectamente compatible con su envío al Centro de Farmacovigilancia y es prioritario que se notifique en el momento en que se produce la reacción. Esto permitiría que las autoridades sanitarias dispusieran de esa información meses antes de su publicación, lo que podría contribuir a proporcionar información para la toma de decisiones si en un momento dado fuera necesario.
Los editores de las revistas biomédicas deberían fomentar la publicación de casos de reacciones adversas de la forma más completa posible para que tanto los demás médicos como los profesionales que evalúan estas sospechas de reacciones adversas puedan obtener conclusiones no sesgadas. También se mejoraría la detección precoz de señales de alerta si los editores exigiesen la notificación previa al Centro de Farmacovigilancia como condición para publicar el caso.
Lo conocido sobre el tema
• El programa de notificación espontánea de sospechas de reacciones adversas a medicamentos permite detectar precozmente señales de alerta sobre problemas de seguridad con medicamentos.
• Los casos de sospechas de reacciones adversas a medicamentos publicados en revistas biomédicas españolas se introducen en la base de datos nacional de sospechas de reacciones adversas (FEDRA).
• El número de notificaciones necesario para generar una señal de alerta depende, entre otros factores, de la calidad de la información de las notificaciones.
Qué aporta este estudio
• La calidad de la información de los casos de reacciones adversas a medicamentos publicados en revistas biomédicas no ha mejorado después del envío a los editores de revistas biomédicas españolas de las recomendaciones elaboradas por el Comité Técnico del Sistema Español de Farmacovigilancia.
• Los apartados con un mayor porcentaje de falta de información son el de dosis, las fechas de tratamiento, las fechas de reacción y, como consecuencia, la secuencia temporal.
• Este estudio destaca no sólo la importancia de que se publiquen los casos de sospecha de reacciones adversas a medicamentos que sean de especial interés, sino también que contengan la información completa necesaria para su evaluación.
Presentado como póster en las VI Jornadas de Farmacovigilancia. Madrid 2006.
Correspondencia:
C. Esteban Calvo.
Centro de Farmacovigilancia de la Comunidad de Madrid. Consejería de Sanidad.
P.º Recoletos, 14. 28001 Madrid. España.
Correo electrónico: carmen.esteban@salud.madrid.org
Manuscrito recibido el 25-1-2008.
Manuscrito aceptado para su publicación el 3-3-2008.
