







Caso clínico. Mujer de 59 años, sin antecedentes personales de interés, que acudió a la consulta por presentar un cuadro de astenia y artralgias generalizadas de dos semanas de evolución. En la exploración física se objetiva artritis bilateral de MCF y de rodillas. Ante la sospecha de enfermedad reumática se solicitaron hemograma, PCR, FR y ANAS, que fueron normales, encontrándose una VSG de 55 y en la bioquímica un patrón de colestasis disociada con GOT de 81, GPT de 127, GGT de 132 y FA de 680, por lo que se realizaron serologías a VHA, VHB y VHC y eco abdominal, con resultados normales. Dos semanas después, la paciente presentaba dos nuevos síntomas: tos seca y febrícula vespertina, y en la exploración física destacaban nódulos subcutáneos (de 2 × 2 cm de diámetro) dolorosos a la palpación en ambas piernas, por lo que se solicitó una radiografía de tórax que demostró adenopatías hiliares derechas (figs. 1 y 2). Se planteó el diagnóstico diferencial entre tuberculosis, sarcoidosis y linfoma, y la paciente fue remitida al hospital de referencia para continuar el estudio. Se realizaron un test de Mantoux, que fue negativo, y una TAC torácica en la que se objetivaron adenopatías hiliares bilaterales y mediastínicas, sin alteraciones parenquimatosas ni pleurales. Posteriormente se realizó una biopsia hepática, con el resultado de granulomatosis epitelioide no necrosante. La exploración oftalmológica, la biopsia de los nódulos subcutáneos y la gammagrafía pulmonar fueron normales. Se estableció el diagnóstico de sarcoidosis aguda con afección ganglionar mediastínica e hiliar, hepática y cutánea (eritema nudoso). La paciente fue tratada con prednisona, 30 mg cada 12 horas durante 15 días y posteriormente en pauta descendente. La paciente evoluciona de manera favorable, quedando asintomática tras el tratamiento. En la revisión a los 6 meses del inicio del cuadro, sigue asintomática, apreciándose una mejoría de la colestasis disociada y disminución de las adenopatías hiliares.
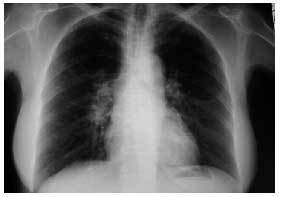
Figura 1. Radiografía de tórax PA: adenopatías hiliares derechas.
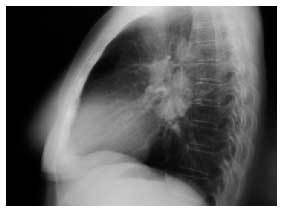
Figura 2. Radiografía de tórax lateral: adenopatías hiliares derechas.
Discusión y conclusiones. La sarcoidosis es una enfermedad granulomatosa multisistémica de etiología desconocida, caracterizada histológicamente por granulomas epitelioides no caseificantes que afectan a diferentes tejidos y órganos1. Afecta fundamentalmente a adultos jóvenes (20-30 años), siendo más frecuente en mujeres y en norteamericanos de raza negra. Hasta el 50% de los afectados es diagnosticado accidentalmente por una radiografía de tórax de rutina. Las características más típicas de la enfermedad son adenopatías hiliares bilaterales, infiltrados pulmonares y lesiones dermatológicas y/o oculares. El órgano más afectado es el pulmón (80-90%), siendo la tríada clínica característica (tos, disnea y dolor torácico)2. Otros síntomas frecuentes son: eritema nudoso, lupus pernio, hepatoesplenomegalia, uveítis, poliartritis, adenopatías periféricas, parálisis del nervio facial, alteraciones cardíacas, neurológica, óseas, etc.3,4. La radiografía de tórax está alterada hasta en el 90% de los pacientes (desde adenopatías hiliares bilaterales hasta fibrosis pulmonar). También son frecuentes el patrón restrictivo, la disminución del test de difusión de CO, el aumento de la VSG, la hipercalciuria, el aumento de GOT, GPT y FA, así como de ECA. La confirmación histológica se realiza mediante biopsia de tejidos afectados (si no existen lesiones periféricas, se harán biopsia transbronquial o mediastinoscopia). Otras pruebas diagnósticas son TAC torácico, TAC con galio 67 y lavado broncoalveolar5. El diagnóstico diferencial se plantea con la tuberculosis (principal enfermedad a descartar), neumonitis por hipersensibilidad, linfomas, etc. Son necesarias tres condiciones para establecer el diagnóstico de sarcoidosis: clínica y/o anomalías en la radiografía de tórax compatibles, evidencia histológica de granulomas no caseificantes y excluir infecciones y otras enfermedades con histología y clínicas similares. La evolución y el pronóstico se correlacionan con la forma de presentación y la extensión de la enfermedad. Las remisiones espontáneas se dan hasta en dos tercios de los pacientes, las secuelas permanentes en el 10-20% y la mortalidad se sitúa en 1-5%. En todos los pacientes se debería hacer un seguimiento durante los primeros 2 años por el alto porcentaje de recaídas. No existen fármacos que prevengan la aparición de fibrosis pulmonar. Los corticoides aceleran la desaparición de los síntomas y los cambios radiográficos, sin influir en el pronóstico6. Cuando hay afección sistémica, se utilizan corticoides orales a dosis de 20-40 mg/día de prednisona. En algunos casos refractarios al tratamiento con corticoides se utilizan citotóxicos (metotrexato y azatioprina).
