





Introducción. En 1992, Brugada y Brugada describieron un síndrome caracterizado por episodios de síncope y muerte súbita en pacientes sin cardiopatía estructural, con patrón electrocardiográfico de bloqueo de rama derecha y elevación del segmento ST en las derivaciones precordiales V1 a V31. Se han identificado 3 mutaciones del cromosoma 3 que afectan a la función del canal de sodio cardíaco y causan la presentación de trastornos de conducción, con períodos refractarios ventriculares heterogéneos, que facilitan la aparición de arritmias. El electrocardiograma (ECG) puede verse modulado por cambios autonómicos o la administración de antiarrítmicos, y en ocasiones es difícil su diagnóstico debido a la existencia de formas intermitentes u ocultas del síndrome2. Causa un 50% de las muertes súbitas anuales en pacientes jóvenes con corazón normal2. Actualmente su tratamiento consiste en la implantación de un desfibrilador.
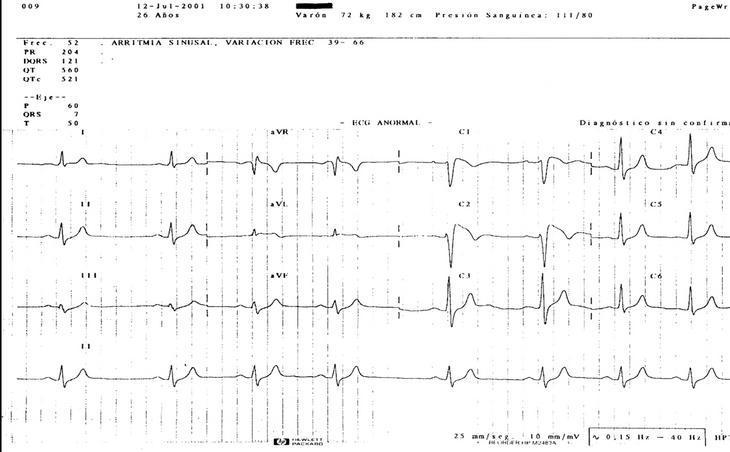
Caso clínico. Varón de 21 años, sin alergias medicamentosas, hábitos tóxicos, ni antecedentes patológicos de interés. El paciente refiere padre con hipertrofia ventricular y hermano y tío paterno muertos a los 18 años de muerte súbita. El paciente acude a urgencias por fiebre de 39 oC, náuseas y palpitaciones. En la exploración destaca: tensión arterial, 140/70 mmHg; frecuencia cardíaca, 104 lat/min y frecuencia respiratoria, 20 resp/min. La auscultación revela soplo sistólico 2/6 mesocárdico, y el resto de la exploración es normal. Se solicita analítica (hemograma, bioquímica y estudio tiroideo), que es normal. El ECG muestra bloqueo incompleto de rama derecha con elevación del segmento ST en precordiales derechas de V1 a V2 (fig. 1). El paciente se deriva al servicio de cardiología donde, después de practicarle una inducción con ajmalina, se le diagnostica el síndrome y se le implanta un desfibrilador.
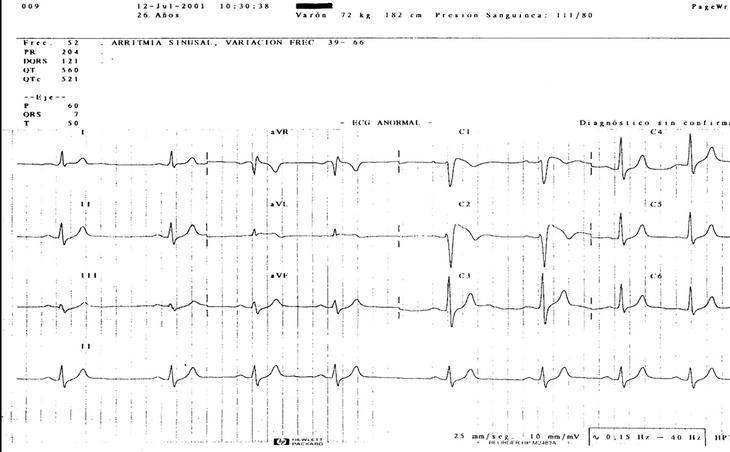
Fig 1
Discusión y conclusiones. En el síndrome de Brugada se han descrito 2 morfologías del segmento ST en precordiales derechas: una convexa (coved) y otra en forma de silla de montar (saddle-back)2. La primera se asocia a un fuerte potencial arritmógeno y la segunda a un curso crónico. En nuestro caso observamos la alternancia de las 2 morfologías. Al revisar la historia del paciente hallamos un ECG, practicado en 1992, cuando aún no se había descrito el síndrome, con morfología tipo saddle-back; otro ECG practicado en 1994 era normal, mientras que el último ECG presentaba una morfología de tipo coved. Este caso demuestra el carácter dinámico de los cambios del ECG, mostrando a veces patrones poco evidentes del síndrome que pueden confundir el diagnóstico. En muchos casos publicados del síndrome, las arritmias se presentan en reposo o durante el sueño, sugiriendo una causa bradicardia-dependiente. La hipótesis de que el bloqueo simpático y la estimulación vagal podrían inducir arritmias ventriculares malignas se apoyaría en la presentación de cambios del ECG por la acción del sistema nervioso autónomo o los antiarrítmicos. Dado que la temperatura corporal puede causar variaciones en la cinética de los canales de sodio, creemos que la hipertermia y el exceso de sudación por el síndrome febril causaron la arritmia y el consiguiente diagnóstico de nuestro paciente.
