



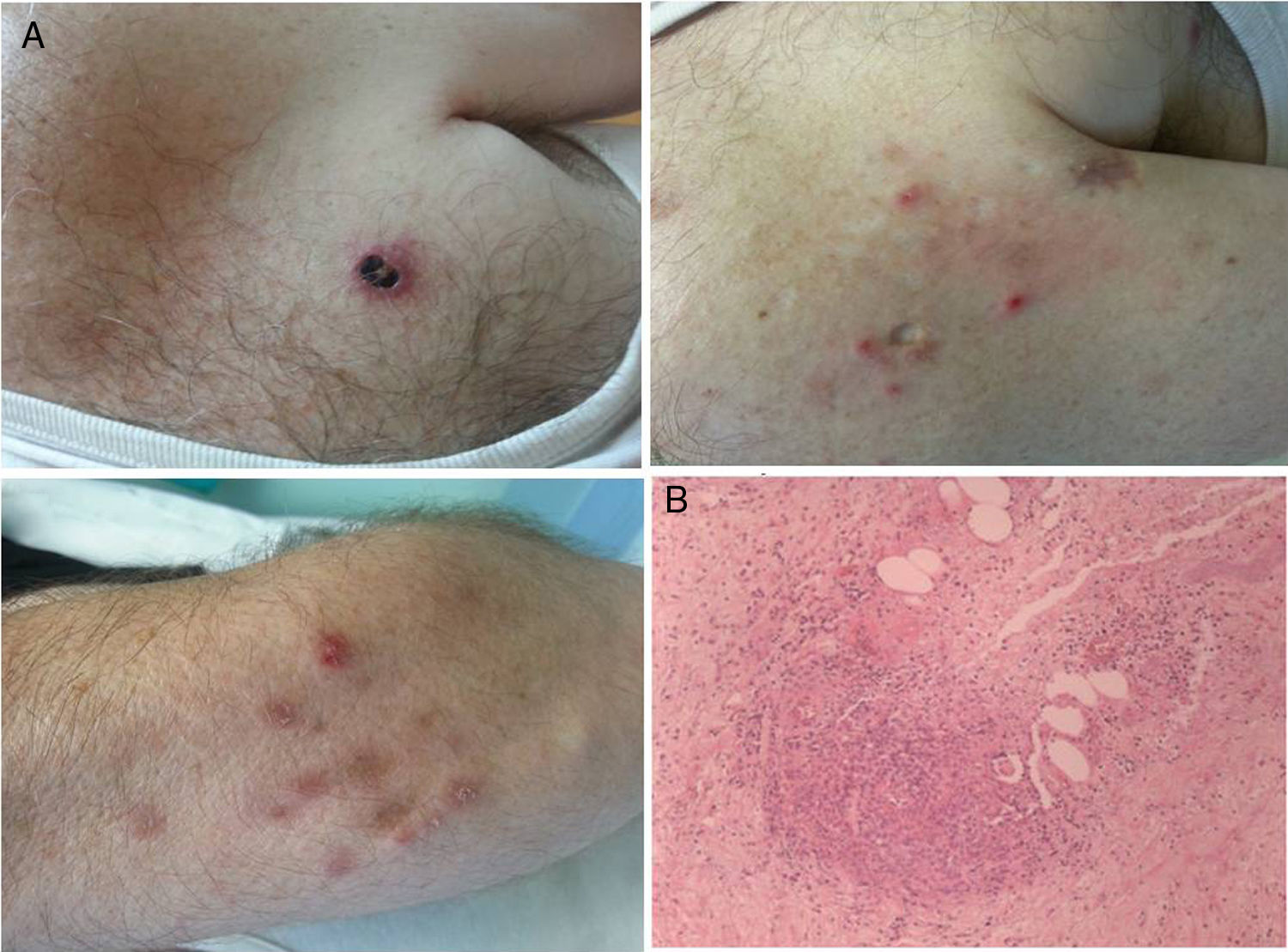
Paciente varón de 56 años, profesor, fumador de 30 cigarros al día y como antecedente de interés, lumbalgias de repetición por discopatía degenerativa en L4-L5 con severa esclerosis.
Hace 13 meses comenzó a notar la presencia de lesiones en la piel, más frecuentes en el tronco, y algunas en las extremidades superiores. Dichas lesiones, acompañadas de prurito ocasional, comenzaron como una roncha o habón de aspecto eccematoide, que llegaban a ulcerarse siendo en ese momento dolorosas, apareciendo posteriormente una costra negruzca y luego curaron, dejando una cicatriz blanquecina con cierta profundidad. Las lesiones se encontraban en diferentes estadios (fig. 1A). El paciente no presentó pérdida de peso, ni fiebre, ni otros síntomas generales.
. Corte biopsia de papulosis linfomatoide (B).")
Las exploraciones básicas fueron completamente normales, sin presencia de adenopatías, masas ni visceromegalias. De entrada se le solicitó analítica básica de salud, que puso de manifiesto un colesterol de 255mg/dl, con LDL de 162mg/dl, PCR de 13,6mg/l y VSG a la 1.a hora de 31mm. Serología para virus y bacterias, negativas. Se remitió a la consulta de dermatología tras tratamiento corticoideo tópico, para valoración biópsica y tratamiento si procedía.
En consulta de oncodermatología se le citó para biopsia. La misma puso de manifiesto un denso infiltrado inflamatorio linfocitario, neutrófilo y eosinófilo a nivel intersticial con degeneración de fibras colágenas, destacando la presencia de linfocitos medianos y grandes de disposición perivascular, en dermis superficial y profunda, que tras el estudio inmunohistoquímico presentaron positividad para CD30 y CD8, con pérdida de CD5 y CD2, siendo negativos para CD56 y EBER (hibridación in situ). Se identificaron además estructuras vasculares trombosadas, asociadas a áreas de necrosis y hemorragia. La epidermis suprayacente se presentó ulcerada e hiperplásica, con espongiosis y exocitosis. No se detectaron microorganismos tras tinción histoquímica para Gram y Giemsa. Todo ello compatible con papulosis linfomatoide (PL) tipo E (fig. 1B).
El estudio de extensión para el diagnóstico diferencial se completó mediante TAC cérvico-torácico-abdómino-pélvico con contraste que solo evidenció un enfisema centroacinar y paraseptal en campos pulmonares superiores.
El tratamiento administrado ha sido metotrexato 10mg a la semana con ácido fólico 5mg al día junto con corticoides tópicos.
La PL es un trastorno linfoproliferativo de linfocitos T CD30+ cutáneo primario. Fue descrita por Macaulay en 1968 como «erupción continua con autorreparación, clínicamente benigna e histológicamente maligna». Es una rara enfermedad cutánea crónica recurrente, y de etiología desconocida1. La PL se incluye en el grupo descrito, junto al linfoma cutáneo anaplásico de células grandes y las lesiones borderline. Hay varios tipos según los rasgos histoquímicos (A, B, C…). Su mecanismo etiopatogénico se ha vinculado con factores genéticos e inmunitarios. La PL es más propia de adultos y ancianos, con predominio en el sexo masculino. El curso clínico suele ser prolongado (varios años) y generalmente benigno, aunque el 10-20% progresa hacia linfomas2. La PL se caracteriza por la presencia de lesiones exclusivamente cutáneas papulonodulares múltiples, con regresión espontánea en semanas y tendencia a recidivar. La anatomía patológica de estas lesiones muestra una infiltración dérmica por linfocitos T atípicos junto a un número variable de células inflamatorias con patrón histoquímico propio3. El diagnóstico diferencial puede a veces resultar muy difícil. El metotrexato es uno de los fármacos utilizados en su abordaje4. Por su potencial maligno incierto, los pacientes deben ser cuidadosamente estudiados, con un seguimiento a largo plazo. Para esto se impone el estudio multidisciplinario, que incluya al médico de familia, dermatólogo, anatomopatólogo y al hematólogo.
