




Congenital malformations of the chest wall comprise a heterogeneous group of diseases denominated spondylocostal dysostosis. They have in common developmental abnormalities in the morphology of the structures of the chest and vertebrae with a broad characterization: from mild deformity without functional consequences to life-threatening injuries. We present the case of a girl with spondylocostal dysostosis and acute cholangitis.
Case reportA 13-month-old female patient with severe malnutrition, history of hydrocephalus and myelomeningocele at birth was admitted in the pediatric emergency room with fever and progressive respiratory distress. Clinical assessment revealed ribs and vertebral malformations and acute cholangitis.
ConclusionsComplex rib abnormalities consist in deformities of the chest wall, which do not have a particular pattern and are extremely rare. When they are associated with myelomeningocele and hydrocephalus, they may be considered as autosomal recessive inheritance spondylocostal dysostosis. The diagnosis is established by clinical assessment and X-rays. Spondylocostal dysostosis identification and complications related to their genetic and molecular causes are still a challenge for the clinical pediatricians and the multidisciplinary medical team who treats these patients throughout lifetime.
Las malformaciones congénitas vertebrales y costales concomitantes comprenden un grupo heterogéneo de enfermedades denominadas disostosis espondilocostal. Tienen en común la alteración del desarrollo o morfología de las estructuras vertebrales y de la caja torácica con una expresividad variable: desde la deformidad leve sin consecuencias funcionales hasta lesiones que amenazan la vida. Se presenta el caso de una niña con disostosis espondilocostal y colangitis aguda.
Caso clínicoPaciente de sexo femenino de 13 meses de edad, con desnutrición severa y antecedente de hidrocefalia y mielomeningocele, quien ingresa al servicio de Urgencias por presentar dificultad respiratoria progresiva y fiebre. En la evaluación, se encontraron malformación costo-vertebral y colangitis aguda.
ConclusionesLas anormalidades costales complejas consisten en malformaciones de la pared torácica sin un patrón determinado y son extremadamente raras. Cuando se presentan al mismo tiempo que las malformaciones vertebrales, puede considerarse como síndrome de disostosis espondilocostal ligado a herencia autosómica recesiva. El diagnóstico es clínico-radiográfico. La identificación de la disostosis espondilocostal y las complicaciones relacionadas con sus causas genético-moleculares implican un reto para el pediatra y el equipo multidisciplinario que los trata a lo largo de su vida.
Chest wall malformations are classified into five types: type I, cartilaginous (pectus excavatum, pectus carinatum); type II, costal (simple, which in turn can be single, double, or combined; and complex: fused or syndromic); type III, chondro-costal (Poland syndrome, thoracopagus); type IV, sternal (sternal cleft); type V, clavicle-scapular (clavicular, scapular, combined). They can be part of syndromes such as spondylocostal dysostosis, and spondylothoracic dysostosis, characterized by rib and spine abnormalities with or without neural tube defects and other malformations, or may appear as isolated defects.1 In patients with spondylocostal dysostosis, multiple defects of vertebral segmentation and costal abnormalities (fusion, reduction in number, misalignment) are observed.
Clinically, a short trunk, short neck, and scoliosis characterize chest wall malformations. The diagnosis is based on the radiographic findings. Subtypes are defined by identifying two mutated alleles in one of the four genes (DLL3, MESP2, LFNG, and HES7)2 in which pathogenic variants are known to be transmitted by autosomal recessive inheritance. Alterations affect the Notch signaling pathway, which is critical for the coordination of this process. Moreover, in patients with autosomal dominant inheritance, alterations in transcription activation of TBX6 protein have been found, probably due to haploinsufficiency.3
2Clinical caseWe present the case of a 13-month-old female patient with ectomorph external habitus, macrocephaly due to hydrocephalus with a functional, right ventriculoperitoneal shunt placed when the patient was 22 days of life (surgical scar 2cm in right iliac fossa). She presented myelomeningocele corrected at birth (surgical scar of 8cm in the sacrococcygeal region). She was the product of a second pregnancy without prenatal control and was born by cesarean at 36.6 weeks of gestation. A 5-year-old sister and parents healthy, who denied consanguinity, drug addiction, chronic or degenerative diseases, exposure to environmental toxicants or the presence of similar lesions in other relatives. When admitted to the emergency room, her weight was 7kg (percentile < 3 according to the World Health Organization tables), height of 63cm (percentile < 3), and cephalic perimeter of 55cm (percentile > 97). Respected muscle tone, decreased tropism, capable of walking with help. Vital signs were the following: heart rate 131/min, 42 breaths/min, axillary temperature 38.5°C, oxygen saturation 97% by pulse oximetry, and blood pressure 94/49mmHg.
The patient was admitted due to the following symptoms: fever, progressive respiratory difficulty, hepatomegaly of 4cm, asymmetry in the thoracic excursion secondary to hypomotility, right hemithorax, mild chest wall depression by palpation, and a discrete protrusion at crying during auscultation. No other clinical or pathological features were observed (Table 1).
Summary of clinical and radiographic elements of the patient.
| Background |
| Hydrocephalus |
| Neural tube defect (corrected myelomeningocele) |
| Physical examination |
| Macrocephaly with hydrocephalus |
| Low weight for age, normal for height |
| Low height for age (diminished thoracic height) |
| Delay in speech and language development, stands up and walk with help |
| Fever |
| Difficulty breathing |
| Right rib cage asymmetry |
| Hepatomegaly |
| Radiographic elements |
| Agenesis of the right first rib |
| Hypoplasia of the left first rib |
| Bilateral aberrant clavicle insertion |
| Sixth and seventh rib arches merged into right hemithorax |
| T6 and T7 butterfly vertebrae |
| Gastromegaly |
| Current disease |
| Cholangitis |
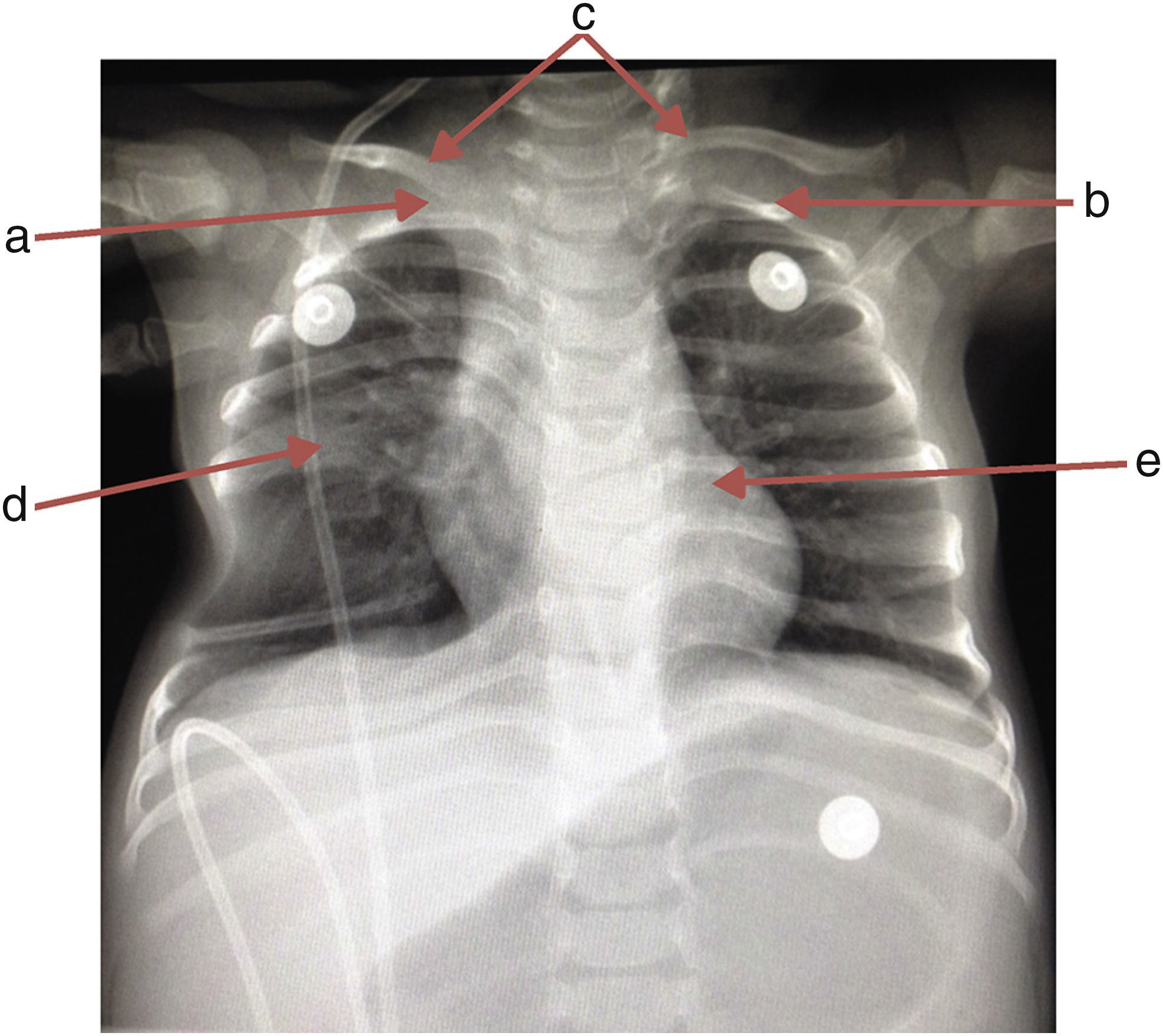
A chest x-ray was requested, which showed the absence of the first right rib and hypoplasia of the first left rib that lead to an aberrant insertion of the clavicles. The sixth and seventh costal arches were merged into the right hemithorax upwards; the inferior costal arches were displaced downwards forming a space that enhanced the transparency of the lung, and T6 and T7 butterfly vertebrae (Fig. 1).
, left first rib hypoplasia (b), aberrant insertion of both clavicles (c), sixth and seventh costal arch fused in the right hemithorax (d) and T6 and T7 butterfly vertebra (e).")
An ultrasound from liver and bile ducts was performed; choledochus with fusiform dilatation of 1.9cm was found near its union with the Wirsung duct. Cholangitis was suspected. An evaluation by a pediatric infectious disease specialist was requested, who suggested the prescription of piperacillin-tazobactam (before blood culture). A multidisciplinary intervention was solicited with the services of Nutrition, Thoracic Surgery, and Pulmonology, who recommended conservative treatment, including medical checkups every three months to evaluate the thoracic and pulmonary development. The Pulmonary Physiology service instructed the mother about pulmonary hygiene measures. The patient was transferred to the Pediatric Surgery service 24hours later, where a laparoscopic cholangiography was performed, and cholangitis was confirmed.
The process aimed at nutritional recovery within the hospital was initiated. Fourteen days later, the patient left asymptomatic and was referred for follow-up.
3DiscussionDuring weeks of gestation three to five, the mesoderm (between the endoderm and ectoderm, both sides of the notochord) differentiates into somites and gives rise to the sclerotome (costal and vertebral processes), the myotome (musculature) and the dermatome (deeper layers of skin and subcutaneous tissue).1,4 The formation of somites from the unsegmented precursor tissue, known as presomitic mesoderm, is tightly controlled at the molecular level by the interaction of several signal-transduction pathways including FGF, Wnt, and Notch. In this process, Notch signaling pathway is activated in the presomitic mesoderm in regular pulses, which leads to the periodic activation of HES7 and LFNG genes.5 Any disorder during this stage of embryogenesis can result in vertebral, costal, and abdominal defects. HOX gene also has been implicated in costal malformation.6
The term spondylocostal dysostosis is used to describe a wide variety of radiological features that include multiple abnormal vertebral segmentation, usually contiguous, and the involvement of malalignment, fusions, and absence of some ribs.5,7–9
Chest wall deformities may present respiratory insufficiency at birth, pass unnoticed or progress to respiratory failure during the patient's development. Therefore, the diagnosis may be delayed until adulthood. The present case corresponds to a type II chest wall deformity (3.2% of the total chest wall deformities). It is associated with uncommon anatomical alterations; each one constitutes a unique variety, whose prognosis, diagnosis, and treatment need to be evaluated accurately. Differential diagnoses of the costal malformations include Poland syndrome,10 the cerebrocostomandibular syndrome, Edwards syndrome,11,12 VACTERL-H syndrome,13 the lumbo-costo-vertebral syndrome,14,15 the spondylothoracic dysostosis (Jarcho-Levin syndrome16,17 and Cassamassima-Morton-Nance syndrome18 with high mortality due to respiratory failure) (Table 2), and spondylocostal dysostosis.1–4
Comparison between differential diagnoses and the present case.
| Differential diagnosis | Year of publication | Clinical features | Present case |
|---|---|---|---|
| Poland syndrome10 Autosomal dominant inheritance | 1841 | • Unilateral aplasia of costal fascicles of the pectoralis major muscle • Lack of minor pectoralis muscle • Breast and nipple hypoplasia • Unilateral brachydactyly • Thoracic wall deformity • Costal hypoplasia | X X X X X √ |
| Cerebrocostomandibular syndrome Both dominant and recessive autosomal inheritance | 1966 | • Microcephaly • Bilateral asymmetric costal anomalies • Micrognathia | X √ X |
| Edwards syndrome11,12 Autosomal recessive inheritance linked to the X chromosome | 1960 | • Costal anomalies (sharp clavicles, narrow ribs) • Vertebral anomalies • Myelomeningocele • Microcephaly • Prominent occiput • Ogival palate • Micrognathia | X √ √ X X X X |
| VACTERL-H syndrome13 Autosomal recessive inheritance | 1972 | There must be three or more abnormalities for the diagnosis: • Vertebral defects • Heart defects • Kidney defects • Limb anomalies • Anal atresia • Tracheoesophageal fistula • Esophageal or duodenal atresia • Hydrocephalus | √ X X X X X X √ |
| Lumbocostovertebral syndrome14,15 Diabetic mothers, lysergic acid exposure, probably autosomal recessive inheritance | 1972 | Costovertebral and chest wall musculature abnormalities: • Hemivertebrae • Absence of ribs • Spinal dysraphism • Anterior myelomeningocele • Hypoplasia of the anterior abdominal wall (a lumbosacral hernia) • Hepatopathy • Arthrogryposis • Congenital heart disease • Renal agenesis | √ √ √ X X X X X X |
| Cassamassima-Morton-Nance syndrome (OMIM 271520)18 Affected genes Pax 1 and Pax 9. Autosomal recessive inheritance, de novo mutations, and chromosomal rearrangements | 1981 | • Segmentation defects • Short thorax • Costal anomalies (fusion) • Asymmetrical vertebrae • Hemivertebrae, vertebral fusion • Short stature, short trunk • Spina bifida/myelomeningocele • Difficulty of breath or respiratory failure • Scoliosis, anal atresia, genitourinary/anal malformation (characteristic) • Dysmorphic craniofacial features, short neck, and low mobility • Cardiac abnormalities • Wide forehead, dolichocephalous, broad nasal bridge, anteverted nares, whistling appearance of the mouth (occasional) • Intellectual deficit (uncommon feature) | √ √ √ √ √ √ √ √ X X X X X |
| Jarcho-Levin syndrome16,17 Autosomal dominant inheritance | 1938 | • Short stature (due to spinal alterations) at the expense of short upper segment • Vertebral anomalies (primary characteristic) • Costal anomalies • Neural tube defects (myelomeningocele) • Hydrocephalus • Difficulty/shortness of breath • Recurrent respiratory infections • Cardiovascular anomalies (septal defects) • Diaphragmatic hernia • Genitourinary anomalies • Meckel diverticulum • Bifid uvula | √ √ √ √ √ √ √ X X X X X |
√, present sign; X, absent sign.
In 1968, Rimoin et al. proposed the term spondylocostal dysplasia for the localized malformations in the spine and ribs. In 1991, Karnes et al., based on radiographic findings, redefined the Jarcho-Levin syndrome in two types: spondylothoracic dysostosis and spondylocostal dysostosis. Mortier et al., in 1996, divided the defects of multiple segmentation into three categories: Jarcho-Levin syndrome (symmetrical thorax with crab-like appearance), spondylothoracic dysostosis and spondylocostal dysostosis; all of them with possible extraskeletal defects.7,17,19,20
Currently, the diagnosis of spondylocostal dysostosis is clinical and radiographic. However, there is a wide variety of imaging phenotypes described, which have been used to describe costal and vertebral abnormalities but also have generated confusion in the nomenclature. For this reason, the International Consortium for Vertebral Anomalies and Scoliosis (ICVAS) proposed an algorithm for ontogenic research. This algorithm simplifies the comparison and stratification of spinal (curvature, length), vertebral (normal, single or multiple segmentation and morphology), rib cage (symmetry, asymmetry, size, shape) and ribs (symmetry, asymmetry, number, fusion) defects.2,4,8
Chest wall deformities accompanied by neural tube defects and short stature secondary to spinal abnormalities are compatible with spondylocostal dysostosis, which is a rare genetic disorder with a prevalence of 0.25/10,000 live births.16 It belongs to a group of hereditary diseases characterized by multiple defects of vertebrae segmentation (hemivertebrae, agenesis, butterfly and hypoplastic vertebrae) with abnormalities of the ribs (fusion, agenesis, misalignment) associated with neural tube defects, such as hydrocephalus and the Arnold-Chiari malformation. Every neurological malformation that accompanies the syndrome is one of its components; for example, the dysgenesis of the corpus callosum, holoprosencephaly, and myelomeningocele (lumbosacral, thoracic or lumbar). Also, there may be other abnormalities such as renal, genitourinary, gastrointestinal, limb, and congenital heart defects.
Skeletal anomalies should be detected on radiographs and ultrasonographic studies (cardiac, abdominal, and renal) to establish a diagnosis. Subsequently, the clinical and radiological findings should be considered to evaluate if they are consistent with any of the disorders included in the differential diagnosis. Also, the family history should be considered, especially the cases of affected individuals or consanguinity of the parents.
Once the diagnosis of spondylocostal dysostosis has been established, the radiographic phenotype is used to determine the possible genes involved.
Spondylocostal dysostosis due to genetic causes has been classified into two groups: the first includes the severe forms of spondylocostal dysostosis, with malformation of ten or more vertebrae, usually linked to an autosomal recessive transmission with complete penetration. It includes mutations in genes DLL3 (SCDI; OMIM 277300), MESP2 (SCD2; OMIM 608681), LFNG (SCD3; OMIM 609813), and HES7 (SCD4; OMIM 613686). This group also includes the phenotypically different spondylothoracic dysostosis syndrome, which is caused by mutations in the MESP2 gene. In the second group, the autosomal dominant form of spondylocostal dysostosis, only some vertebrae are affected. Evidence has shown that it is due to haploinsufficiency with variable penetrance and includes mutations in TBX6. The Klippel-Feil syndrome is caused by mutations in GDF6 (KFSI; OMIM 118100) or GDF3 (KFS3; OMIM 613702), and congenital scoliosis is caused by mutations in MESP2 or HES7.2–4,8 Somitogenesis of HEY and HES genes should be noted since they are involved in the regulation of neurogenesis, vasculogenesis, cardiogenesis, and cancer.
The four types of spondylocostal dysostosis due to autosomal recessive inheritance have distinctive radiographic phenotypes. Better evidence is needed to determine whether genotype correlates with phenotypes 3 and 4. Clinical features include a short trunk and neck, mild scoliosis (generally non-progressive), defects of cost-vertebral segmentation, among others.
Spondylocostal dysostosis 1 (associated with DLL3 gene) involves four diagnostic criteria plus an irregular pattern of vertebral bodies ossification on spinal radiographs prenatally and in early childhood. Each vertebral body has a round or ovoid shape with smooth boundaries (pebble beach sign). The main affectation is usually located on the thorax. Consanguinity has been reported in 75% of the cases.
In spondylocostal dysostosis 2 (associated with MESP2), all vertebral segments show at least some disruption to form and shape; the lumbar vertebrae are the most affected. Consanguinity has been reported.
In spondylocostal dysostosis 3 (associated with LFNG), shortening of the spine is more severe than types 1 and 2. All bodies appear to show more serious segmentation defects. Rib anomalies are similar to those observed in types 1 and 2. It resembles sexually transmitted diseases and has been reported in only one family.
Spondylocostal dysostosis 4 (associated with HES7) resembles spondylothoracic dysostosis with severe vertebral segmentation anomalies. The vertebral pedicles are relatively prominent (“tramline” sign) compared with type 1. Cases in two families in southern Europe have been reported. There is damage of chromosome 17p13.1 (MIM 613686 phenotype) by HES7 mutation (gene/locus MIM 608059), which has been associated with defects in laterality and neural tube formation.4,5
Spondylothoracic dysostosis, despite the similarities to autosomal recessive spondylocostal dysostosis, presents individual phenotypic characteristics (Table 3).21 It has been described in people with sexually transmitted diseases and gene MESP2 affection. Furthermore, it has been widely described by Cornier et al. in Puerto Rican individuals with Spanish ascendance.22 This phenotype is currently known as Jarcho-Levin syndrome. However, when it is accompanied by imperforate anus, genitourinary malformations, and other extraskeletal malformations, it is called the Cassamassima-Morton-Nance syndrome.20
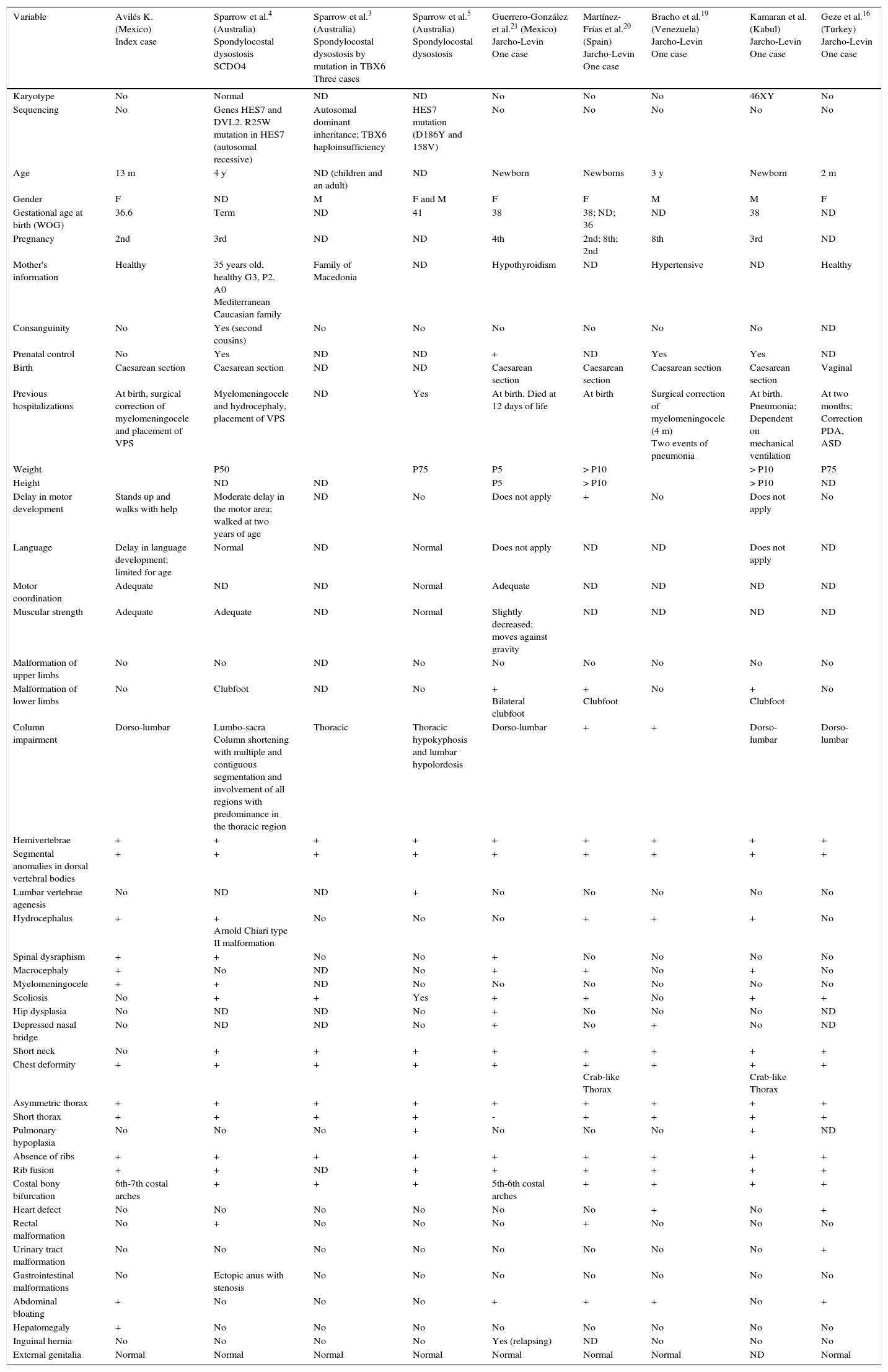
Comparison of the index case with other cases reported in the literature with costal-vertebral malformations due to spondylocostal dysostosis and spondylothoracic dysostosis.
| Variable | Avilés K. (Mexico) Index case | Sparrow et al.4 (Australia) Spondylocostal dysostosis SCDO4 | Sparrow et al.3 (Australia) Spondylocostal dysostosis by mutation in TBX6 Three cases | Sparrow et al.5 (Australia) Spondylocostal dysostosis | Guerrero-González et al.21 (Mexico) Jarcho-Levin One case | Martínez-Frías et al.20 (Spain) Jarcho-Levin One case | Bracho et al.19 (Venezuela) Jarcho-Levin One case | Kamaran et al. (Kabul) Jarcho-Levin One case | Geze et al.16 (Turkey) Jarcho-Levin One case |
|---|---|---|---|---|---|---|---|---|---|
| Karyotype | No | Normal | ND | ND | No | No | No | 46XY | No |
| Sequencing | No | Genes HES7 and DVL2. R25W mutation in HES7 (autosomal recessive) | Autosomal dominant inheritance; TBX6 haploinsufficiency | HES7 mutation (D186Y and 158V) | No | No | No | No | No |
| Age | 13 m | 4 y | ND (children and an adult) | ND | Newborn | Newborns | 3 y | Newborn | 2 m |
| Gender | F | ND | M | F and M | F | F | M | M | F |
| Gestational age at birth (WOG) | 36.6 | Term | ND | 41 | 38 | 38; ND; 36 | ND | 38 | ND |
| Pregnancy | 2nd | 3rd | ND | ND | 4th | 2nd; 8th; 2nd | 8th | 3rd | ND |
| Mother's information | Healthy | 35 years old, healthy G3, P2, A0 Mediterranean Caucasian family | Family of Macedonia | ND | Hypothyroidism | ND | Hypertensive | ND | Healthy |
| Consanguinity | No | Yes (second cousins) | No | No | No | No | No | No | ND |
| Prenatal control | No | Yes | ND | ND | + | ND | Yes | Yes | ND |
| Birth | Caesarean section | Caesarean section | ND | ND | Caesarean section | Caesarean section | Caesarean section | Caesarean section | Vaginal |
| Previous hospitalizations | At birth, surgical correction of myelomeningocele and placement of VPS | Myelomeningocele and hydrocephaly, placement of VPS | ND | Yes | At birth. Died at 12 days of life | At birth | Surgical correction of myelomeningocele (4 m) Two events of pneumonia | At birth. Pneumonia; Dependent on mechanical ventilation | At two months; Correction PDA, ASD |
| Weight | P50 | P75 | P5 | > P10 | > P10 | P75 | |||
| Height | ND | ND | P5 | > P10 | > P10 | ND | |||
| Delay in motor development | Stands up and walks with help | Moderate delay in the motor area; walked at two years of age | ND | No | Does not apply | + | No | Does not apply | No |
| Language | Delay in language development; limited for age | Normal | ND | Normal | Does not apply | ND | ND | Does not apply | ND |
| Motor coordination | Adequate | ND | ND | Normal | Adequate | ND | ND | ND | ND |
| Muscular strength | Adequate | Adequate | ND | Normal | Slightly decreased; moves against gravity | ND | ND | ND | ND |
| Malformation of upper limbs | No | No | ND | No | No | No | No | No | No |
| Malformation of lower limbs | No | Clubfoot | ND | No | + Bilateral clubfoot | + Clubfoot | No | + Clubfoot | No |
| Column impairment | Dorso-lumbar | Lumbo-sacra Column shortening with multiple and contiguous segmentation and involvement of all regions with predominance in the thoracic region | Thoracic | Thoracic hypokyphosis and lumbar hypolordosis | Dorso-lumbar | + | + | Dorso-lumbar | Dorso-lumbar |
| Hemivertebrae | + | + | + | + | + | + | + | + | + |
| Segmental anomalies in dorsal vertebral bodies | + | + | + | + | + | + | + | + | + |
| Lumbar vertebrae agenesis | No | ND | ND | + | No | No | No | No | No |
| Hydrocephalus | + | + Arnold Chiari type II malformation | No | No | No | + | + | + | No |
| Spinal dysraphism | + | + | No | No | + | No | No | No | No |
| Macrocephaly | + | No | ND | No | + | + | No | + | No |
| Myelomeningocele | + | + | ND | No | No | No | No | No | No |
| Scoliosis | No | + | + | Yes | + | + | No | + | + |
| Hip dysplasia | No | ND | ND | No | + | No | No | No | ND |
| Depressed nasal bridge | No | ND | ND | No | + | No | + | No | ND |
| Short neck | No | + | + | + | + | + | + | + | + |
| Chest deformity | + | + | + | + | + | + Crab-like Thorax | + | + Crab-like Thorax | + |
| Asymmetric thorax | + | + | + | + | + | + | + | + | + |
| Short thorax | + | + | + | + | - | + | + | + | + |
| Pulmonary hypoplasia | No | No | No | + | No | No | No | + | ND |
| Absence of ribs | + | + | + | + | + | + | + | + | + |
| Rib fusion | + | + | ND | + | + | + | + | + | + |
| Costal bony bifurcation | 6th-7th costal arches | + | + | + | 5th-6th costal arches | + | + | + | + |
| Heart defect | No | No | No | No | No | No | + | No | + |
| Rectal malformation | No | + | No | No | No | + | No | No | No |
| Urinary tract malformation | No | No | No | No | No | No | No | No | + |
| Gastrointestinal malformations | No | Ectopic anus with stenosis | No | No | No | No | No | No | No |
| Abdominal bloating | + | No | No | No | + | + | + | No | + |
| Hepatomegaly | + | No | No | No | No | No | No | No | No |
| Inguinal hernia | No | No | No | No | Yes (relapsing) | ND | No | No | No |
| External genitalia | Normal | Normal | Normal | Normal | Normal | Normal | Normal | ND | Normal |
m, months; y, years; WOG, weeks of gestation; VPS, ventriculoperitoneal shunt; P, percentile for age; ND, not described; +/-, element present/absent; PDA, patent ductus arteriosus; ASD, atrial septal defect.
Regarding acute biliary tract infections in the absence of biliary atresia, it is known that they are rare in children (0.13%-0.22%).23 This acute and severe condition due to the bile duct inflammation and infection is defined as cholangitis, and it is characterized by hepatalgia, fever, and jaundice (Charcot triad). Acute cholangitis is a systemic disease with high mortality, for which the medical treatment is urgent.24 The present case is the first reported case of a child with spondylocostal dysostosis and acute cholangitis; the latter is a risk factor that complicates the preexisting condition.
In cases with spondylocostal dysostosis, Teli et al. have reported that the conservative treatment with chest physiotherapy increases the survival rate. The most serious complication is the respiratory failure. Surgical treatment is reserved for no responsive children: those that require rib cage stabilization or have spinal deformities such as progressive scoliosis.9
After the comprehensive evaluation of the case, the classification of the radiographic phenotype according to the vertebral and costal malformations, the history of myelomeningocele and hydrocephalus plus an extensive literature review, we determined that the case corresponds to spondylocostal dysostosis type 4. No description was found to explain the presence of acute bacterial cholangitis in a female pediatric patient with no risk factors described, such as bile duct malformations.
Complex costal malformations are infrequent. Therefore, if they are accompanied by vertebral abnormalities and neural tube defects, spondylocostal dysostosis syndrome should be considered. The radiographic phenotype is essential to diagnose the subtype. The diagnosis is necessary to establish the treatment to prevent respiratory failure, and other intrathoracic, vertebral and extraskeletal development complications, and subsequently, a multidisciplinary follow-up. However, the classification of spondylocostal dysostosis and its variants is still complicated; thus, it remains a challenge for the pediatrician and the multidisciplinary team treating these patients throughout their lifetime.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe author declares no conflicts of interest of any nature.
We thank Dr. Eloy López-Marure, pediatric radiologist from the Radiology and Imaging Service of the Juan I. Menchaca Civil Hospital, Guadalajara, Jalisco, Mexico; and Dr. Hugo Gutiérrez-González, resident of Pediatrics at the Fray Antonio Alcalde Civil Hospital, Guadalajara, Jalisco, Mexico.
Please cite this article as: Avilés-Martínez KI. Disostosis espondilocostal y colangitis aguda en urgencias pediátricas. Bol Med Hosp Infant Mex. 2016;73:256–267.