


La leucoencefalopatía con sustancia blanca evanescente es una de las leucodistrofias más frecuentes. Generalmente inicia en la infancia y presenta un patrón de herencia autosómica recesiva. El 90% de los casos manifiesta mutaciones en uno de los genes que codifican para las cinco subunidades del factor de iniciación eucariótica 2 (EIF2B5). El diagnóstico se realiza por las manifestaciones clínicas, hallazgos en la resonancia magnética cerebral y estudios moleculares confirmatorios.
Caso clínicoPaciente masculino de 13 meses con neurodesarrollo previo normal. Antecedente de internamiento por vómito, hipertermia, irritabilidad y rechazo a la vía oral de 15 días de evolución. Ante la exploración presentó perímetro cefálico y pares craneales normales. Se encontró hipotónico, con reflejos incrementados, sin datos meníngeos ni de cráneo hipertensivo. La tomografía de cráneo mostró hipodensidad generalizada de la sustancia blanca. Egresó sin recuperar deambulación. A los 15 días presentó somnolencia y crisis convulsivas focales después de traumatismo craneoencefálico. En la resonancia magnética se observó hipointensidad generalizada de sustancia blanca. Ante la sospecha de leucoencefalopatía con sustancia blanca evanescente, se solicitó la secuenciación del gen EIF2B5, que reportó mutación homocigota c.318A>T en el exón 2. El paciente requirió múltiples hospitalizaciones por hipertermia y descontrol de crisis convulsivas. Posteriormente mostró deterioro cognitivo, motor y pérdida de la agudeza visual. Falleció a los 6 años por neumonía severa.
ConclusionesEste caso contribuye a conocer el espectro de mutaciones que se presenta en pacientes mexicanos y permite ampliar el fenotipo asociado con esta mutación.
Vanishing white matter disease is one of the most frequent leukodystrophies in childhood with an autosomal recessive inheritance. A mutation in one of the genes encoding the five subunits of the eukaryotic initiation factor 2 (EIF2B5) is present in 90% of the cases. The diagnosis can be accomplished by the clinical and neuroradiological findings and molecular tests.
Case reportWe describe a thirteen-month-old male with previous normal neurodevelopment, who was hospitalized for vomiting, hyperthermia and irritability. On examination, cephalic perimeter and cranial pairs were normal. Hypotonia, increased muscle stretching reflexes, generalized white matter hypodensity on cranial tomography were found. Fifteen days after discharge, he suffered minor head trauma presenting drowsiness and focal seizures. Magnetic resonance showed generalized hypointensity of white matter. Vanishing white matter disease was suspected, and confirmed by sequencing of the EIF2B5 gene, revealing a homozygous c.318A> T mutation in exon 2. Subsequently, visual acuity was lost and cognitive and motor deterioration was evident. The patient died at six years of age due to severe pneumonia.
ConclusionsThis case contributes to the knowledge of the mutational spectrum present in Mexican patients and allows to extend the phenotype associated to this mutation.
La leucoencefalopatía con sustancia blanca evanescente (VWM, por sus siglas en inglés leukoencephalophaty with vanishing white matter; OMIM#603896), también conocida como ataxia infantil con hipomielinización, es una de las leucodistrofias más frecuentes en población caucásica1,2. El primer caso fue descrito por W. Eicke, en 1962, en una mujer de 36 años con amenorrea secundaria, que después de traumatismos craneocencefálicos leves, desarrolló deterioro neurológico progresivo hasta su fallecimiento3.
La VWM es una enfermedad hereditaria con un patrón de transmisión autosómica recesiva, que en el 90% de los casos se asocia con mutaciones en alguno de los cinco genes EIF2B1 (12q24.3), EIF2B2 (14q24), EIF2B3 (1p34.1), EIF2B4 (2p23.3), EIF2B5 (3q27) encargados de codificar las cinco subunidades (α, β, γ, δ, ¿) que conforman el factor de iniciación de la traducción eucariótica 2 (eIF2B)4,5. Su principal función es iniciar la traducción de ARN mensajeros a polipéptidos y su regulación en estados de estrés. El factor se activa por ciclos de fosforilación y desfosforilación acoplada a guanosín difosfato (GDP)/guanosín trifosfato (GTP) en su subunidad GEF (guanine nucleotide exchange factor). Estados de estrés celular que promueven la fosforilación de eIF2α inhiben la función reactivadora de eIF2B; su baja concentración intracelular limita los ciclos de reactivación, disminuyendo la síntesis global de proteínas6. Aunque se encuentra distribuido en diferentes líneas celulares, las células gliales presentan mayor vulnerabilidad al estrés mientras las neuronas y otras células del sistema nervioso central (SNC) están protegidas. Esta vulnerabilidad lleva a la alteración en la producción de la mielina, característica del padecimiento7,8.
En 1997, van der Knaap y colaboradores definieron la VWM en función de criterios clínicos y neurorradiológicos8. La forma prenatal de la enfermedad presenta oligohidramnios y disminución de los movimientos fetales; al momento del nacimiento, los pacientes manifiestan dificultad para la alimentación, vómito, hipotonia, crisis convulsivas de difícil control, estado de coma, apneas, microcefalia, cataratas, visceromegalias, hipoplasia renal, disgenesia ovárica, contracturas articulares y muerte antes de cumplir un año de edad9. La forma infantil se manifiesta en el primer año de vida con irritabilidad, estupor, crisis convulsivas, neurorregresión motora, infecciones recurrentes; usualmente, los pacientes fallecen antes de los 2 años de edad10–12. La forma infantil tardía presenta dos variedades: unos pacientes desarrollan ataxia, temblor de intención y dismetría de 1 a 5 años de edad, con un periodo previo de desarrollo motor y cognitivo normal, algunos con retraso motor y de lenguaje leve. Otros casos muestran coma posterior a traumatismo craneoencefálico (TCE) o fiebre, y aunque hay recuperación, los pacientes desarrollan deterioro motor progresivo con problemas de la marcha, datos de neurona motora superior, disartria, crisis convulsivas, problemas en la alimentación y atrofia de nervio óptico. En estos pacientes, parece que las habilidades cognitivas están preservadas, pero posteriormente entran en una fase de estabilización hasta que fallecen en un periodo de 1 a 5 años8,13. La forma juvenil inicia entre los 5 y 15 años de edad, con diplejía espástica de lenta progresión que alterna con periodos de estabilidad, e incluso de mejoría motora; los pacientes tienen una sobrevida larga14. En la forma del adulto predomina el deterioro cognitivo, episodios transitorios de neuritis óptica, hemiparesia, cefalea intensa. En mujeres se presenta amenorrea secundaria por falla ovárica referida como una ovarioleucodistrofia15,16. En publicaciones recientes, se ha hecho énfasis en la presencia de trastornos del movimiento, como distonía y mioclonus, que caracterizan a las formas tardías, pero ocasionalmente se han descrito en formas infantiles con opsoclonus-mioclonus17.
Las anormalidades neurorradiológicas son constantes, pero varían según la edad, y pueden estar presentes hasta en pacientes asintomáticos. El estudio de elección es la resonancia magnética (RM) de cráneo. En las formas neonatales solamente se observa inmadurez caracterizada por giros gruesos y la sustancia blanca con alto contenido de agua. En las formas infantiles, la sustancia blanca hemisférica está afectada de forma simétrica y difusa en secuencias T1, T2 y FLAIR (recuperación de inversión atenuada de fluido) con una intensidad de señal similar a la de líquido cefalorraquídeo (LCR). También se aprecia una malla fina de tejido restante dentro de las áreas afectadas, con apariencia radiada, y en el centro semioval se detecta un patrón punteado en cortes sagitales y coronales del T1 y FLAIR; en esta etapa no se observa atrofia. En las formas tardías es común la presencia de lesiones quísticas observadas en la secuencia FLAIR. La espectroscopia de estas zonas muestra semejanzas con el LCR, con disminución de los metabolitos normales y presencia de lactato y glucosa. La atrofia cerebelosa varía de leve a grave; el vermis es la primera estructura afectada y se pueden observar alteraciones de señal en el mesencéfalo, puente y ocasionalmente en la médula. En adultos con progresión lenta de la enfermedad, se puede observar atrofia córtico-subcortical1,2,8.
En este trabajo, se describe un caso con la forma infantil de VWM por una mutación c.318A>T homocigota en el exón 2 del gen EIF2B5. El caso contribuye a conocer el espectro de mutaciones que se presenta en pacientes mexicanos, lo que permite ampliar el fenotipo asociado con esta mutación.
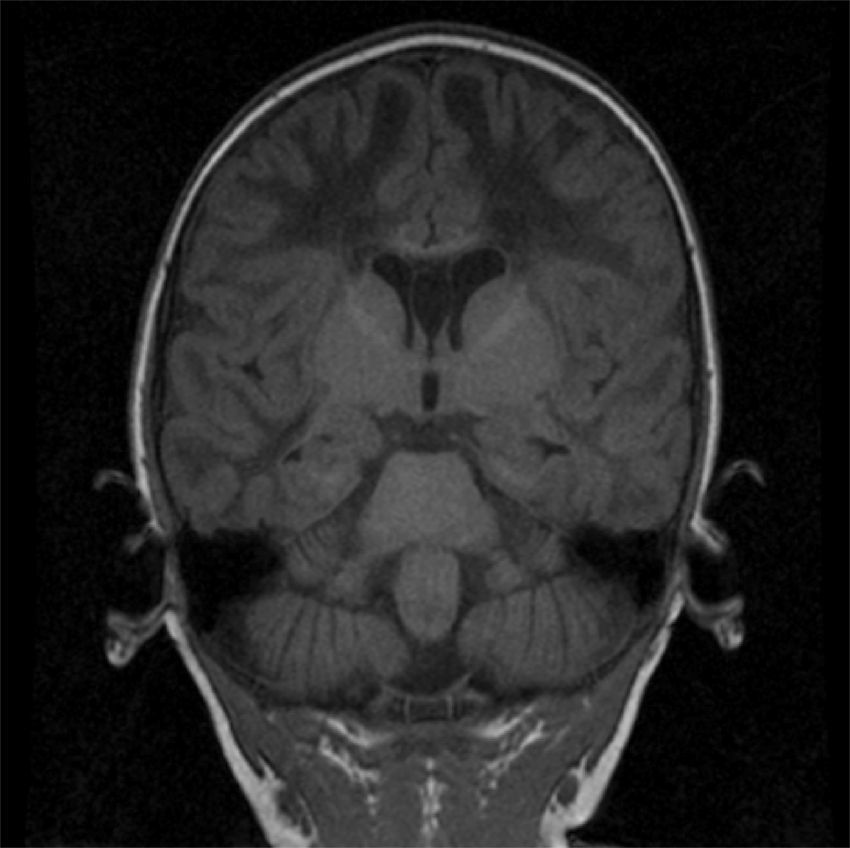
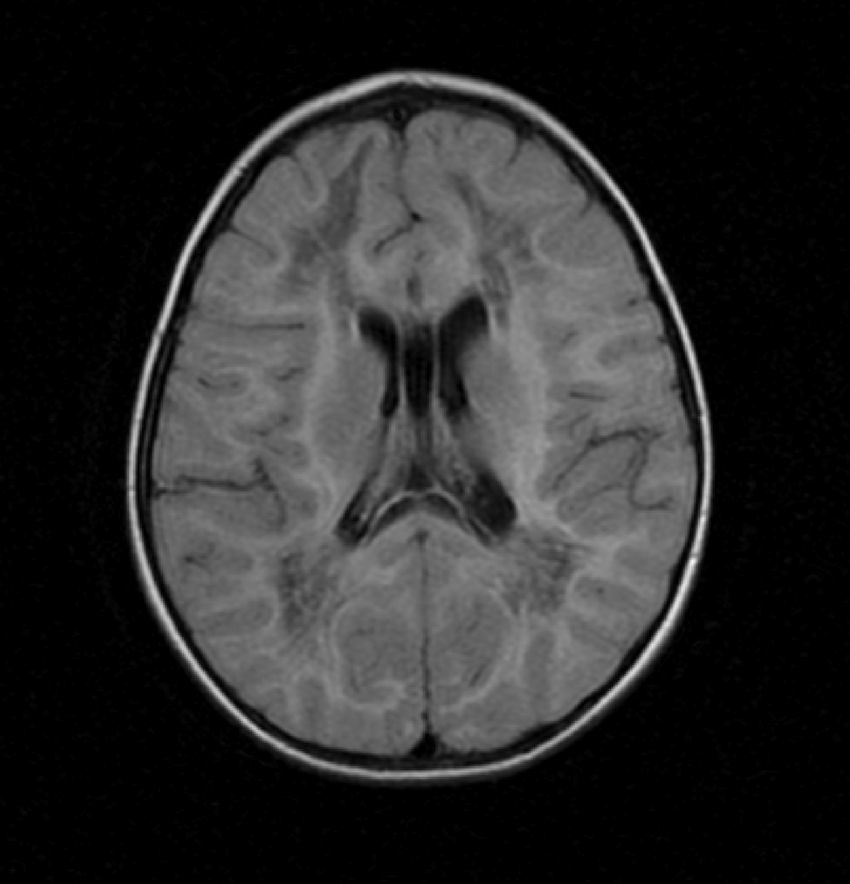
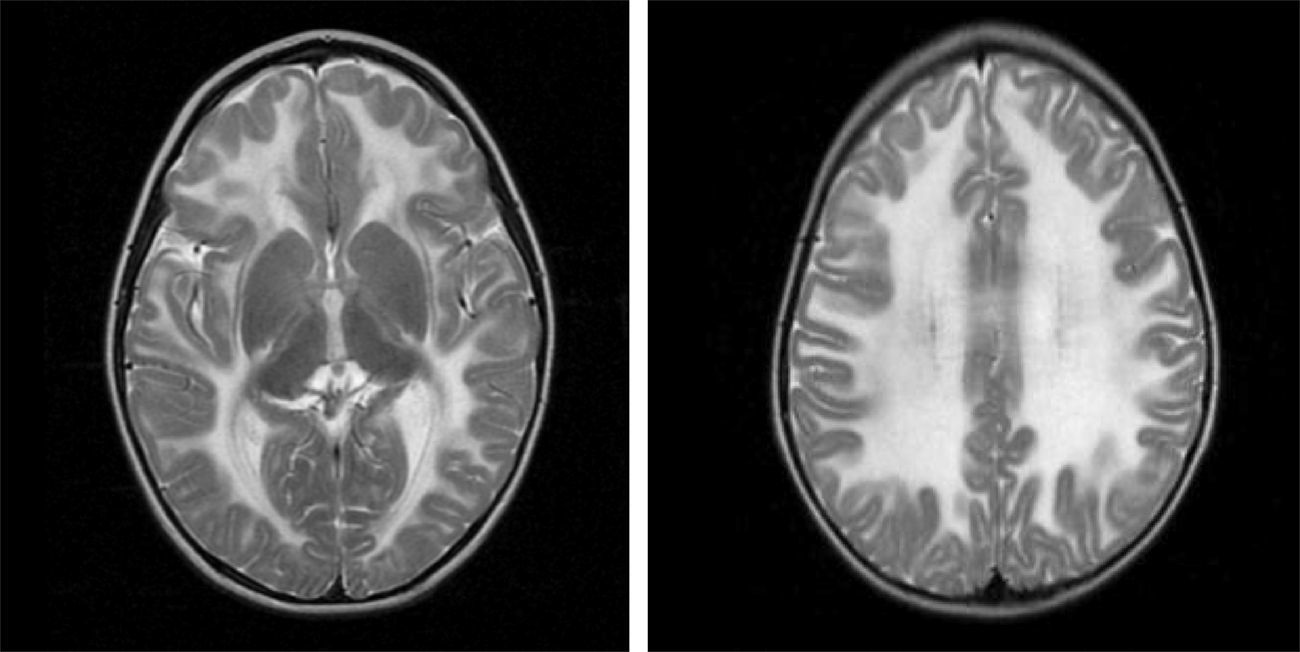
2Caso clínicoPaciente masculino de 13 meses, padres jóvenes, sanos, no consanguíneos. Producto de la primera gesta, de término, sin datos de asfixia perinatal, adecuado desarrollo psicomotor. Acudió a urgencias por vómito, hipertermia intermitente, irritabilidad, rechazo a la vía oral durante quince días. A su ingreso presentaba hipertermia y periodos de somnolencia con irritabilidad. A la exploración por Neuropediatría se encontró irritable, perímetro cefálico normal, pares craneales y fondo de ojo normal, extremidades con disminución del tono y reflejos de estiramiento muscular incrementados, sin datos meníngeos ni de cráneo hipertensivo, abdomen sin visceromegalias y piel sin manchas anormales. La tomografía de cráneo mostró hipodensidad generalizada de la sustancia blanca. La RM de cráneo evidenció alteración en la intensidad de señal de la sustancia blanca en ambos hemisferios cerebrales en las imágenes potenciadas en T1 y T2 (figs. 1-3). Las fibras “U” se encontraron respetadas y había restos de sustancia blanca en las porciones centrales, sobre todo en los centros semiovales, coronas radiadas a nivel periatrial y frontal (figs. 2 y 3). En T1 se observó atrofia del cuerpo calloso con evidencia de hipomielinización (fig. 1). En T2, la corteza cerebral se observó discretamente atrofiada, mientras que los ganglios basales y el tallo cerebral estaban conservados (fig. 3).



Imágenes axiales T2. Alteración en la mielinización de prácticamente toda la materia blanca de ambos hemisferios cerebrales, respetando la sustancia gris, aunque se observa adelgazamiento de la corteza por atrofia. Se detectan algunas imágenes restantes de materia blanca en los centros semiovales y coronas radiadas.
Se descartó leucodistrofia metacromática por estudio enzimático normal. El paciente egresó sin haber recuperado la marcha. En los quince días posteriores presentó crisis convulsivas focales complejas asociadas con fiebre, electroencefalograma anormal paroxístico focal; inició tratamiento con valproato de magnesio. Después de una semana presentó TCE leve. Desarrolló mayor deterioro motor y regresaron los periodos de somnolencia con irritabilidad, vómito e hipotonía. Se sospechó VWM y se solicitó secuenciación del gen EIF2B5. Se reportó mutación c.318A>T homocigota en el exón 2, provocando un cambio de una leucina por una fenilalanina en la posición 106 de la proteína (p.Leu106Phe). Se confirmó el estado heterocigoto en ambos padres. Los estudios moleculares se realizaron en el laboratorio de la VU University Medical Center, en Ámsterdam.
Las hospitalizaciones por hipertermia continuaron, con episodios de descontrol de crisis convulsivas y exacerbaciones de deterioro cognitivo. A los 2 años, al paciente se le colocó sonda de gastrostomía por dificultad en la deglución. En su última evaluación, a los 6 años, mostró crisis convulsivas ocasionales controladas con valproato de magnesio y topiramato, pérdida completa de la agudeza visual por atrofia de nervio óptico y espasticidad en las cuatro extremidades. El paciente recibió terapia física, ortesis, baclofen vía oral y aplicación de toxina botulínica. Finalmente, falleció a los 6 años por neumonía severa adquirida en la comunidad.
3DiscusiónLas enfermedades que afectan la sustancia blanca son un reto en el ámbito de la genética y la neurología pediátrica. Poseen un espectro clínico amplio, con edades de inicio que van desde el nacimiento hasta la edad adulta. Con el fin de facilitar el diagnóstico de estas enfermedades, se les ha clasificado en dos grupos: las formas clásicas, aquellas condiciones hereditarias que afectan principalmente las células gliales, con o sin afección de la mielina del sistema nervioso periférico; son un grupo de cerca de treinta entidades. El segundo grupo son las leucoencefalopatías genéticamente determinadas, que afectan de forma secundaria a la sustancia blanca por afección de las neuronas o por lesiones vasculares o patologías sistémicas. Entre las leucodistrofias, una de las más frecuentes en la infancia es la VWM18,19.
El caso que se reporta inició en la infancia temprana, con características clínicas que evolucionaron de acuerdo con lo descrito para este padecimiento, episodios de deterioro neurológico asociados con TCE y fiebre. En concordancia con este fenómeno, se ha observado que, en modelos animales, los oligodendrocitos normales muestran hiperactivación de cinasas como GCN2, PERK, PKR y HRI, que fosforilan a eIF2, lo que inhibe a eIF2B, restringe la actividad de GEF y disminuye su capacidad de reactivación sin afectar sus funciones. La deficiencia de eIF2B incrementa la señalización de PERK, que al interactuar con eIF2B mutado, inhibe la síntesis de proteínas en los oligodendrocitos y altera su función en condiciones de estrés20–22.
En pacientes con deterioro neurológico agudo durante o después de un proceso febril, se debe pensar en encefalomielitis diseminada aguda o una encefalitis infecciosa. Cuando el deterioro es subagudo o crónico, las enfermedades mitocondriales, como la deficiencia de piruvato deshidrogenasa, piruvato carboxilasa y el síndrome de Aicardi-Goutières, entran al diagnóstico diferencial. En la adrenoleucodistrofia ligada al cromosoma X, al igual que la VWM, la pérdida neurológica se relaciona con TCE. La atrofia de nervio óptico se observa en enfermedad de Canavan, xantomatosis cerebrotendinosa y en trastornos peroxisomales2.
El análisis de las manifestaciones clínicas, y principalmente neurorradiológicas, permitió distinguir la VWM de estos trastornos dismielinizantes. Los hallazgos de la RM en este paciente son compatibles con los criterios de neuroimagen para VWM, ya que la dismielinización afecta ambos hemisferios cerebrales, muestra restos de mielina a nivel de centros semiovales, coronas radiadas, lóbulos frontales y regiones periatriales, compromiso del cuerpo calloso y discreta atrofia de la corteza cerebral2. Se espera que el electroencefalograma al inicio sea normal, y progresivamente se enlentecen los ritmos de base con paroxismos multifocales. Los potenciales evocados visuales y auditivos son normales al inicio, y van incrementando la latencia hasta desaparecer. A diferencia de la leucodistrofia metacromática, la velocidad de conducción y la electromiografía son normales23.
Los principales hallazgos neuropatológicos en VWM son la degeneración quística, gliosis reactiva, astrocitos de morfología anormal y escasez de mielina. Inicialmente, Dietrich y colaboradores asociaron la pérdida de sustancia blanca con la disfunción del astrocito y sus células progenitoras por ausencia de expresión de la proteína ácida fibrilar glial (GFAP) mediada por el ARN de interferencia del gen EIF2B524. Posteriormente, Dooves y colaboradores encontraron acúmulo de ácido hialurónico, que inhibe la proliferación del astrocito durante la gliosis. La afección a células gliales se extiende a estructuras como cerebelo y retina, explicando la disfunción cerebelosa y visual en algunos pacientes, incluido el presente caso25.
Se han reportado más de 160 mutaciones en las cinco subunidades del complejo eIF2B: el 64.7% en el gen eIF2B5, el 17% en eIF2B4 seguido del 15% en eIF2B2. En eIF2B5, la mayoría son sustituciones de aminoácidos, como p.Arg113His, que es la más frecuente en la forma del adulto4. La mutación en este paciente es la segunda en frecuencia y consiste en un cambio de leucina por fenilalanina en la posición 106. Fue reportada inicialmente en estado heterocigoto por Leegwater y colaboradores26. Posteriormente se reportó otro caso heterocigoto en una niña con ataxia y hemiparesia27.
En la literatura se han descrito otros dos casos homocigotos cuya evolución fue menos rápida y grave23. Algunas mutaciones, como la p.Val309Leu, muestran de forma consistente un fenotipo grave mientras que el estado homocigoto para la mutación p.Thr91Ala puede presentarse en adultos asintomáticos y también en casos tempranos26. La mutación p.Arg113His se localiza cercana a la que este caso presentó, con una evolución clínica menos severa, reflejando que no existe una correlación clara entre el efecto de la mutación sobre la actividad de intercambio GEF de eIF2B y la severidad de las manifestaciones clínicas28.
Se identificaron dos casos de pacientes mexicanos con VWM después de la revisión bibliográfica: uno, que inició a los 14 meses sin estudio molecular, y otro con anormalidad homocigota p.Arg299His (c.896G>A) en el gen EIF2B5. En ambos, las manifestaciones fueron muy similares al paciente reportado. Los pacientes mostraron neurodesarrollo previo normal, deterioro motor y cognitivo asociado con un proceso infeccioso, sin recuperación completa de los hitos perdidos. En el segundo caso, el paciente mostró nuevo deterioro posterior a TCE29,30. La descripción del presente caso contribuye al conocimiento de la epidemiologia clínica y molecular del padecimiento. Probablemente, el hecho de que ambos padres sean portadores, sin evidencia de consanguinidad o endogamia, refleje una frecuencia importante de la mutación en México.
En la actualidad no hay tratamiento específico para VWM, solamente intervenciones para las crisis convulsivas, espasticidad y problemas para la alimentación. Se recomienda evitar situaciones de estrés que provoquen deterioro, como infecciones, fiebre y deportes de contacto, aunque no son suficientes para prevenir la progresión de la enfermedad2.
La VWM es una de las leucodistrofias más frecuentes con casos distribuidos en todo el mundo. En nuestro país, probablemente se encuentra subdiagnosticada, específicamente las formas tardías y atípicas del padecimiento.
La sospecha diagnóstica se sustenta en el cuadro clínico. Los hallazgos de neuroimagen suelen ser característicos, pero los estudios de ADN contribuyen en la confirmación diagnóstica. El caso que se reporta presenta una mutación previamente descrita, con algunas discrepancias clínicas entre los individuos ya descritos, por lo que todavía no es posible concluir respecto de la correlación fenotipo-genotipo de la anormalidad genética. Al no contar con un tratamiento particular, el asesoramiento genético y la explicación a los familiares de la historia natural de la enfermedad son importantes en el abordaje clínico. Al confirmarse el estado de portador heterocigoto sano en ambos padres, existe un riesgo de recurrencia de 25% en cada embarazo. En este caso, las opciones reproductivas incluyen el diagnóstico preimplantación, el diagnóstico prenatal, el donador de gametos de uno de los progenitores y la adopción.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoNinguno.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la Dra. Merjo van der Knaap por su apoyo para la realización del estudio molecular.