


La incontinencia pigmenti es una enfermedad genética rara ligada al cromosoma X, letal en el varón, que afecta a todos los tejidos derivados del ectodermo, como piel, faneras, ojos, dientes y sistema nervioso central, y presenta alteraciones de grado variable en la inmunidad celular. Se caracteriza por la disminución de la melanina en la epidermis y su incremento en la dermis.
Caso clínicoSe presenta el caso de una lactante de dos meses de edad con incontinencia pigmenti grave, confirmada con estudio histopatológico de piel, que cursó con alteraciones neurológicas severas y crisis convulsivas. Además, presentó inmunodeficiencia celular grave que condicionó el desarrollo de infecciones que le ocasionaron la muerte.
ConclusionesSe resalta la importancia del diagnóstico clínico temprano, así como la importancia del manejo multidisciplinario de las alteraciones neurológicas y de las complicaciones infecciosas.
Incontinentia pigmenti is a rare, X-linked genetic disease and affects all ectoderm-derived tissues such as skin, appendages, eyes, teeth and central nervous system as well as disorders of varying degree of cellular immunity characterized by decreasing melanin in the epidermis and increase in the dermis. When the condition occurs in males, it is lethal.
Case reportWe present the case of a 2-month-old infant with severe incontinentia pigmenti confirmed by histological examination of skin biopsy. The condition evolved with severe neurological disorders and seizures along with severe cellular immune deficiency, which affected the development of severe infections and caused the death of the patient.
ConclusionsThe importance of early clinical diagnosis is highlighted along with the importance of multidisciplinary management of neurological disorders and infectious complications.
La incontinencia pigmenti, incontinencia pigmentaria o síndrome de Bloch-Sulzberger es una rara enfermedad genética multisistémica ligada al cromosoma X, letal en el varón, que afecta a todos los tejidos derivados del ectodermo, como piel, faneras, ojos, dientes y sistema nervioso central, así como alteraciones de grado variable en la inmunidad celular. Su nombre está dado por la disminución o ausencia de melanina en las células basales de la epidermis y el incremento de la misma en la dermis, por lo que las lesiones adoptan un aspecto blanquecino. El hallazgo histopatológico de la tercera fase de la enfermedad es precisamente la incontinencia o salida del pigmento a la dermis1. La primera descripción sobre la incontinencia pigmenti (IP) fue realizada en Inglaterra en 1906 por Garrod. Sin embargo, la enfermedad fue caracterizada clínicamente en 1920 por Bardach, Bloch, Siemens y Sulzberger, aunque solamente los nombres de Bloch y Sulzberger aparecen en el epónimo de la enfermedad.
La mayoría de los casos de incontinencia pigmenti se producen en forma esporádica como resultado de una mutación de novo consistente en la deleción de gran parte del gen NEMO (Nuclear Factor Kappa B Essential Modifier [NF-κB essential modulator] o IKKγ [IκB kinase-γ]), que incluye los exones del 4 al 10 y que se encuentra en el locus Xq28. Este gen controla la expresión de algunas citocinas que protegen a la célula contra la apoptosis inducida por el TNFα2,3. Los varones que heredan el gen mutado generalmente no sobreviven. Se han descrito pocos casos de varones que han sobrevivido, los cuales se han relacionado con la presencia de un cromosoma X extra o con mosaicismo somático. Las mujeres afectadas sobreviven debido a la presencia de dos cromosomas sexuales X (dicigosidad del cromosoma X) y a la inactivación temprana del cromosoma X que porta la mutación4,5. Recientemente, la IP se ha relacionado con la presencia de alelos hipomórficos; es decir, mutaciones en el gen que no anulan totalmente la función de la proteína que codifica. Estas mutaciones hipomórficas originan una variante alélica de la IP denominada displasia ectodérmica hipohidrótica asociada con inmunodeficiencia severa (ADE-ID). La inmunodeficiencia que presentan los pacientes puede ser hipogammaglobulinemia con niveles bajos de IgG, hipergammaglobulinemia IgM (fenotipo hiper-IgM) o disgammaglobulinemia, debido a que la mutación del gen NEMO genera una señalización deficiente vía CD40, una molécula que activa a los linfocitos, ocasionando una respuesta defectuosa a antígenos polisacáridos y una elevada susceptibilidad a infecciones bacterianas graves. También se ha descrito que NEMO participa en las vías de señalización para la citotoxicidad celular por los NK, lo que puede causar infecciones virales recurrentes6–8.
Se considera que la tasa de morbilidad de la IP es de 0.7 por cada 100,000, con una frecuencia de presentación de aproximadamente 1 por cada 50,000 recién nacidos vivos, y solo constituye el 1% de todos los trastornos neuroectodérmicos.
En México existen pocos datos sobre la incidencia de la IP. En una revisión del Instituto Nacional de Pediatría se diagnosticaron 36 pacientes en 20 años, lo cual representa alrededor de 1.8 casos nuevos por año9,10.
A pesar de que la incontinencia pigmenti es una enfermedad sistémica, la principal manifestación clínica y siempre constante es la afección cutánea, la cual cursa generalmente por cuatro estadios evolutivos:
- 1.
Lesiones eritematosas, vesiculares y en ocasiones pustulosas, especialmente en extremidades y tronco, que generalmente aparecen desde el nacimiento o en las primeras semanas de vida, aunque en algunos casos se han desarrollado in útero.
- 2.
Lesiones verrugosas, queratósicas, entre la segunda y sexta semanas de vida, en tronco, miembros y dedos de los pies. Pueden persistir durante meses y desaparecer sin dejar secuelas.
- 3.
Lesiones hiperpigmentadas que habitualmente aparecen a partir del cuarto mes de vida siguiendo las líneas de Blaschko. Suele ser la etapa más persistente y acompañarse de alopecia. Pueden desaparecer en la segunda o en la tercera década de la vida.
- 4.
Máculas blanquecinas hipomelánicas de forma rayada en las piernas que pueden ser la única manifestación de la IP en el adulto. En esta última etapa además se presentan signos de atrofia de la piel11–17.
La expresión clínica de la incontinencia pigmenti es muy variable, incluso en los miembros de una misma familia afectada. Algunas pacientes solamente presentan alteraciones cutáneas y dentales, mientras que otras pueden tener un curso grave y progresivo con infecciones mortales; en otros casos desarrollan severa incapacidad neurológica y oftalmológica.
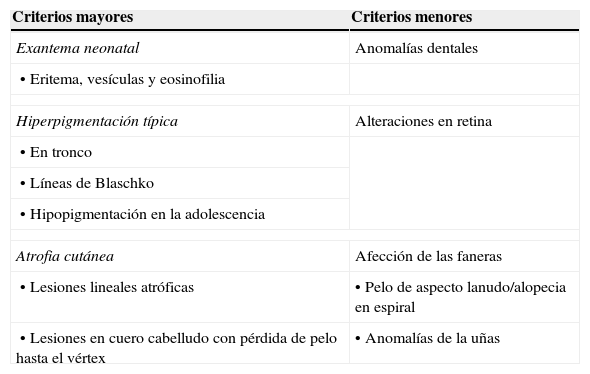
El diagnóstico clínico de incontinencia pigmenti se puede establecer de acuerdo con los criterios propuestos por Landy y Donnai12 (tabla 1). El 50-80% de los casos se acompaña de alteraciones extradermatológicas. La mayoría de las pacientes tienen displasia dental, como hipodoncia, anodoncia, erupción retrasada, coronas en forma cónica, cúspides supernumerarias, anomalías del esmalte y dientes retenidos. El 40% de las pacientes presentan alteraciones oculares, como estrabismo, hipopigmentación retiniana, retinopatía proliferativa, cataratas y manchas corneales. Las alteraciones neurológicas se observan en más de la mitad de los casos, pero solamente el 7.5% con manifestaciones neurológicas graves; el 40% presenta crisis convulsivas que se manifiestan desde la primera semana de la vida. Los estudios de neuroimagen con resonancia magnética nuclear (RMN) pueden mostrar leucomalacia periventricular, hipoplasia del cuerpo calloso, heterotopia de la sustancia gris, anormalidades en cerebelo, encefalomalacia, infartos cerebrales y anormalidades vasculares, y en ocasiones se pueden encontrar lesiones desmielinizantes sin repercusión clínica. Se ha sugerido que las alteraciones oftalmológicas y neurológicas son secundarias a un proceso microangiopático con isquemia secundaria en un momento de alta vulnerabilidad del neonato y que la degranulación de eosinófilos secundaria a la activación de eotaxina genera daños directos en los tejidos. Por lo tanto, los cambios observados en la vasculatura retiniana pueden servir como un marcador potencial de la enfermedad en el sistema nervioso central (SNC). Otras alteraciones descritas en pacientes con IP son hipoplasia mamaria, fibrosis endomiocárdica, insuficiencia tricuspídea e hipertensión pulmonar18–26.
Criterios de clasificación de incontinencia pigmenti. Se requiere de al menos un criterio mayor para el diagnóstico y cualquiera de los menores
| Criterios mayores | Criterios menores |
|---|---|
| Exantema neonatal | Anomalías dentales |
| • Eritema, vesículas y eosinofilia | |
| Hiperpigmentación típica | Alteraciones en retina |
| • En tronco | |
| • Líneas de Blaschko | |
| • Hipopigmentación en la adolescencia | |
| Atrofia cutánea | Afección de las faneras |
| • Lesiones lineales atróficas | • Pelo de aspecto lanudo/alopecia en espiral |
| • Lesiones en cuero cabelludo con pérdida de pelo hasta el vértex | • Anomalías de la uñas |
Ref. Landy y Donnai12
Con respecto a las alteraciones en la inmunidad, se sabe que NEMO (NF-κB essential modulator) es una molécula intracelular que participa en la activación celular por la vía de NF-κB, la cual se activa en la respuesta inflamatoria por algunas citocinas, como TNF-α e IL-1β, y por diferentes productos de microorganismos que reconocen los receptores de la inmunidad innata.
El gen NEMO está involucrado en el desarrollo y en la función del sistema inmune. Es una molécula intracelular que responde rápidamente ante una gran variedad de estímulos involucrados tanto en la respuesta inmune innata como en la adaptativa, como los receptores tipo Toll, receptores que tienen que ver con la muerte celular programada o por daño (NOD/CARD), receptores de la superfamilia de IL-1 y TNF, receptores de IL-15, receptores de IL-17, y los receptores de las células T y B. La activación de NEMO producirá la expresión de una variedad de genes involucrados en la inmunidad, como las proteínas de fase aguda, citocinas, factores de crecimiento, adhesión y coestimulación de moléculas de superficie celular, y reguladores de la proliferación y apoptosis celular.
Las manifestaciones descritas de forma clásica en pacientes con inmunodeficiencia y mutaciones en NEMO son infecciones graves bacterianas en vías aéreas, piel, tejidos blandos, hueso, tracto gastrointestinal, SNC y sepsis, siendo los agentes causales bacterias Gram positivas, como Streptococcus pneumoniae y Staphylococcus aureus, seguido de bacterias Gram negativas, principalmente Pseudomonas spp. y Haemophilus influenzae, además de micobacterias.
Las mutaciones en NEMO (E315a y R319q) asociadas con el síndrome de susceptibilidad mendeliana a las infecciones micobacterianas (MSMD por sus siglas en inglés) tienen un fenotipo celular donde la vía microbio-dependiente se encuentra intacta. Es decir, la sangre de esos pacientes es normal cuando se activa por BCG y citocinas como IL-12 e IFN-γ; en cambio, cuando los mononucleares se activan por PHA o CD3, vía T-dependiente, existe un defecto en la producción de IL-12 y de manera secundaria de IFN-γ. En cultivos celulares de monocitos y linfocitos T activados por la PHA o CD3, el defecto es evidente en los monocitos ya que no pueden producir IL-1227,28. Estas mutaciones en NEMO producen una señalización deficiente en la activación por la vía de CD40-CD40L, ya que según un modelo de la proteína NEMO, las mutaciones producirían una ruptura del puente salino que sirve para que interactúen con la vía CD40. La respuesta al tratamiento farmacológico o combinado con IFN-γ en pacientes con estas mutaciones en NEMO es variable y los pacientes presentan frecuentemente infecciones por micobacterias.
Debido a la mutación referida en el gen NEMO, las células afectadas son refractarias a la estimulación dependiente de NF-kB, por lo tanto son altamente susceptibles a la apoptosis inducida por TNF, sugiriendo así que esta citocina está principalmente involucrada en la aparición de lesiones en IP y asociada también con otras anomalías en la respuesta inmune29–32.
En los hallazgos de la biometría hemática se reporta leucocitosis y eosinofilia del 5 al 79% de los pacientes. Los niveles más altos se observan entre la tercera y quinta semana de vida, coincidiendo con la fase vesicular33–37.
El estudio histopatológico de las lesiones de la piel confirma el diagnóstico, donde los hallazgos histológicos dependerán de la fase en la cual se realice la biopsia.
- •
En la primera fase se observan vesículas intraepidérmicas, espongiosis eosinofílica, células disqueratósicas individuales y remolinos de células escamosas con queratinización central. En la dermis hay infiltrado inflamatorio con predominio de eosinófilos y escasas células mononucleares.
- •
En la segunda fase se observan hiperqueratosis, acantosis y papilomatosis. La queratinización intraepidérmica —evidenciada por los remolinos de células escamosas con queratinización central y la disqueratosis— es más pronunciada que en la fase inflamatoria. En la dermis se observa escaso infiltrado inflamatorio crónico y algunos melanófagos.
- •
En la tercera fase de la enfermedad, la histopatología evidencia epidermis normal o levemente acantótica y en la dermis papilar se observan abundantes macrófagos cargados de melanina o melanófagos.
- •
En la cuarta fase se observa una disminución de la pigmentación de la capa basal y en la dermis los anexos cutáneos están atróficos o ausentes38,39.
Lactante del sexo femenino de tres meses de edad hija de padres de 37 años de edad, no consanguíneos. Sin embargo, ambos procedían de una comunidad pequeña y compartían un mismo apellido, por lo cual existía la posibilidad de endogamia. Una hermana finada a los dos meses de edad por probable sepsis. Tres hermanas sanas. Producto de la gesta V de curso normal obtenida por parto vaginal en la semana 38 de la gestación. Peso al nacer de 3,400g, talla 50cm, no recuerda el Apgar pero se refiere que lloró y respiró al nacer. Egresó junto con la madre a las 24h.
A los ocho días de vida presentó fiebre, rinorrea y tos, por lo que acudió al hospital de su localidad donde se diagnosticó infección de vías respiratorias superiores y eritema facial. A los 15 días, con hiperpigmentación en el dorso de la nariz que se extendió a mejillas, tórax, manos y pies.
A los 18 días de vida presentó un evento de neumonía con insuficiencia respiratoria, por lo cual ingresó en un hospital de su localidad donde presentó sepsis, enterocolitis necrosante, crisis convulsivas, anemia y trombocitopenia. Debido a la mala evolución clínica y persistencia de fiebre, se trasladó al Hospital Infantil de México Federico Gómez.
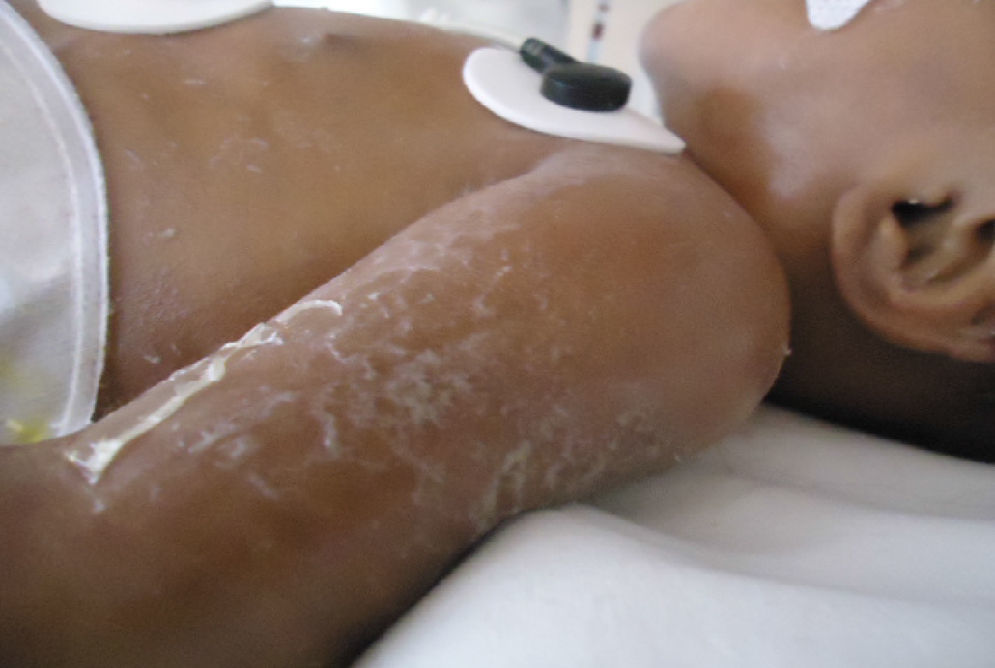
En el momento de su ingreso se encontraba en malas condiciones generales. Al examen físico presentó datos de hipoperfusión tisular. La piel de consistencia dura y acartonada mostraba hiperpigmentación generalizada de predomino en cara y tórax, genitales sin virilización ni macrogenitosomía, con espasticidad en las extremidades (fig. 1).

Los exámenes de laboratorio iniciales mostraron hemoglobina 7.6g/dl, leucocitos 40,600/mm3, neutrófilos 70%, bandas 3%, linfocitos 2%, monocitos 24%, eosinófilos 0%, basófilos 0%, plaquetas 25,000/mm3, glucosa 3mg/dl, BUN 10mg/dl, creatinina 0.4mg/dl, ácido úrico 3.6mg/dl, sodio 122 mEq/l, potasio 2.9 mEq/l, cloro 86 mEq/l, calcio 8.0mg/dl, fósforo 3.3mg/dl, bilirrubina total 1.56mg/dl, bilirrubina directa 0.34mg/l, albúmina 3.4g/dl, ALT 28 U, AST 61 U, GGT 71 U, DHL 592 U, fosfatasa alcalina 136 U, TP 12.8”, TTP 44.7”, INR 0.99, fibrinógeno 259mg/dl.
La gasometría arterial resultó con pH 7.51, pCO2 47.1, pO2 131, HCO3 37.4, lactato 1 mmol/l. Inicialmente se manejó como un cuadro de sepsis nosocomial con foco neumónico mediante la administración de líquidos intravenosos, soporte ventilatorio e inotrópico, doble esquema de antibiótico con cefepime y amikacina. Además, ante la hiperpigmentación de la piel con estado de choque e hiponatremia, se abordó como una crisis adrenal probablemente secundaria a hiperplasia suprarrenal congénita. Sin embargo, los niveles de hormona adrenocorticotropa (ACTH) 25.3 pg/ml, cortisol 13.4μg/dl, androstenediona 312 pg/dl, testosterona < 20 ng/dl, DHE-S 20.4μg/dl, se encontraron dentro de rangos normales con excepción del cortisol que se encontró elevado por el proceso infeccioso.
Se realizó una punción lumbar que reportó normal el estudio citoquímico del líquido cefalorraquídeo (LCR). El hemocultivo inicial resultó positivo para Klebsiella pneumoniae, multisensible. Los resultados de los estudios serológicos para virus herpes tipo I y II, CMV, VIH, rubéola y VDRL se reportaron negativos.
El ecocardiograma se reportó con retornos venosos sistémicos y pulmonares normales, concordancia AV y VA, FE 72%, FA 37% y la PSVD 37mm Hg. El examen oftalmológico mostró fondo de ojo normal y leucoma corneal izquierdo. El estudio de EEG mostró actividad lenta generalizada en rango delta pulipunta y ondas agudas entremezcladas en hemisferio derecho.
La tomografía de cráneo mostró aumento del espacio subaracnoideo a nivel frontal. Parénquima cerebral con adecuada relación de sustancia gris-blanca sin datos de lesiones ocupantes. Hiperintensidad a nivel de ganglios de la base y tálamos compatible con sangrado o infarto cerebral. La RMN de cráneo con contraste con imágenes hipointensas en los núcleos basales en las secuencias T1 y T2. Adelgazamiento del cuerpo calloso en todos sus segmentos, ensanchamiento del espacio subaracnoideo de forma generalizada.
El aspirado de médula ósea reportó celularidad disminuida, megacariocitos ausentes, promielocitos 2%, mielocitos 15%, metamielocitos 14%, bandas 10%, segmentados 6%, eosinófilos 0%, monocitos 2%, linfocitos 45%, basófilos 2%, normoblastos 2% y blastos 2%. Inmunoglobulinas séricas: IgA 35.1mg/dl (6-58mg/dl), IgG 671mg/dl (270-780mg/dl), IgM 26.2mg/dl (12-87mg/dl), IgE 16.9 UI/ml (0-100 UI/ml).
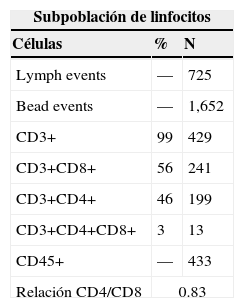
Subpoblación de linfocitos mediante citometría de flujo en sangre periférica (tabla 2). La última biometría hemática mostró hemoglobina 9.3g/dl, leucocitos 1,000/mm3, neutrófilos 34.4% (340), linfocitos 48.9% (490), monocitos 0.16% (16), eosinófilos 0.9% (10), basófilos 0%, plaquetas 45,000/mm3.
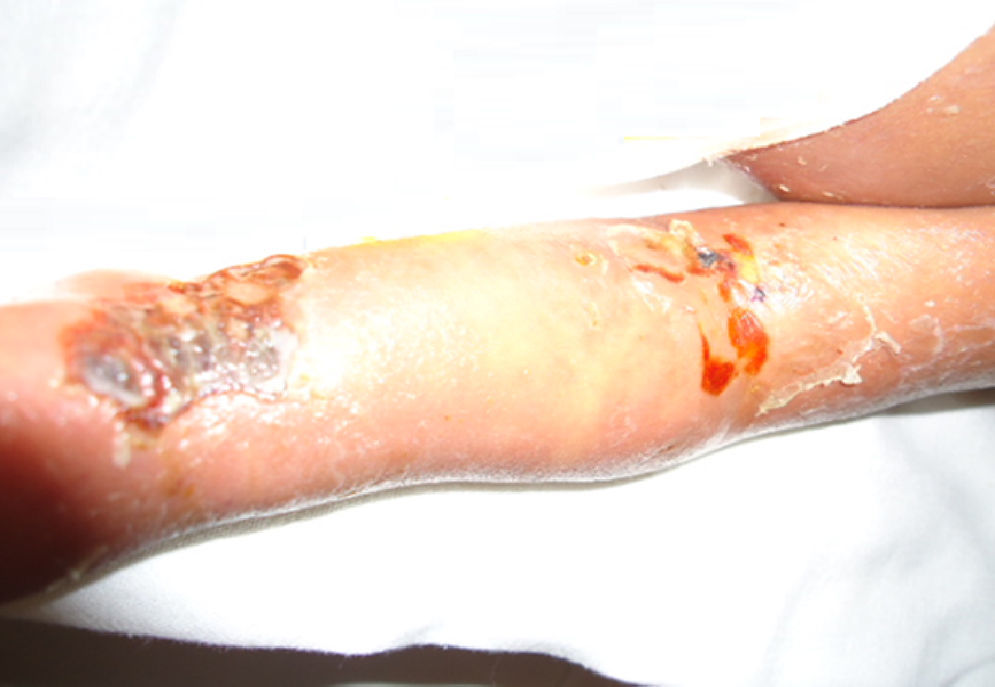
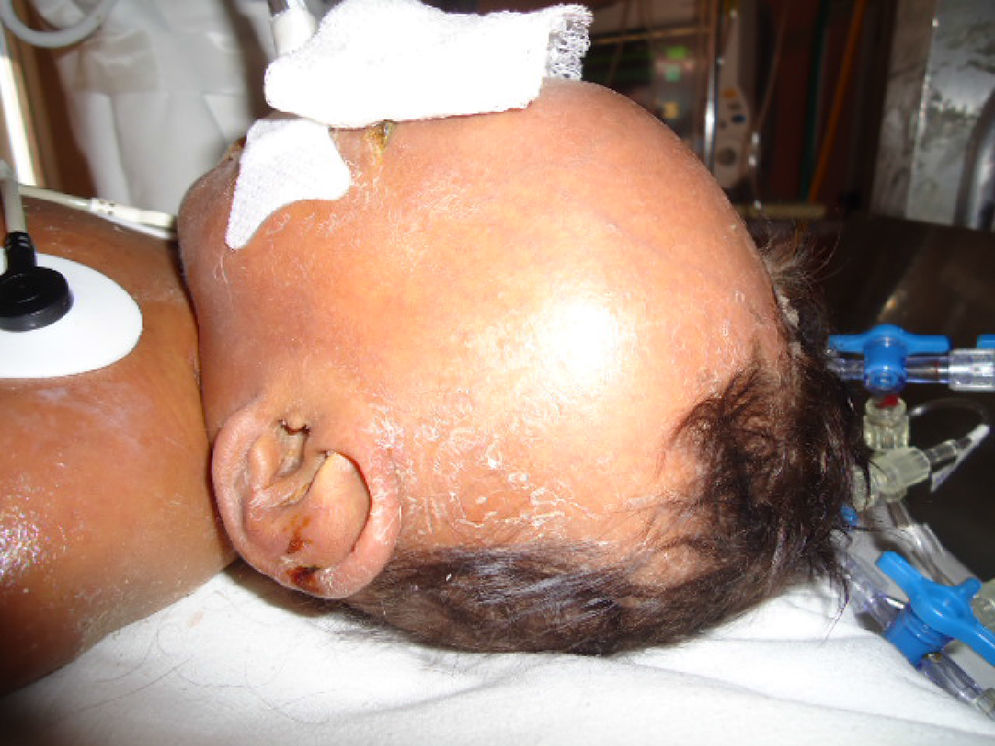
Durante su estancia presentó exantema maculopapular seguido de formación de vesículas y ampollas en pliegues de flexión de las extremidades (fig. 2), que al romperse formaban placas verrugosas con hiperqueratosis y cambios en la pigmentación de la piel del tronco; también se observó alopecia en la región frontal hasta el vértex (fig. 3). Se manejó con cremas humectantes y emolientes.


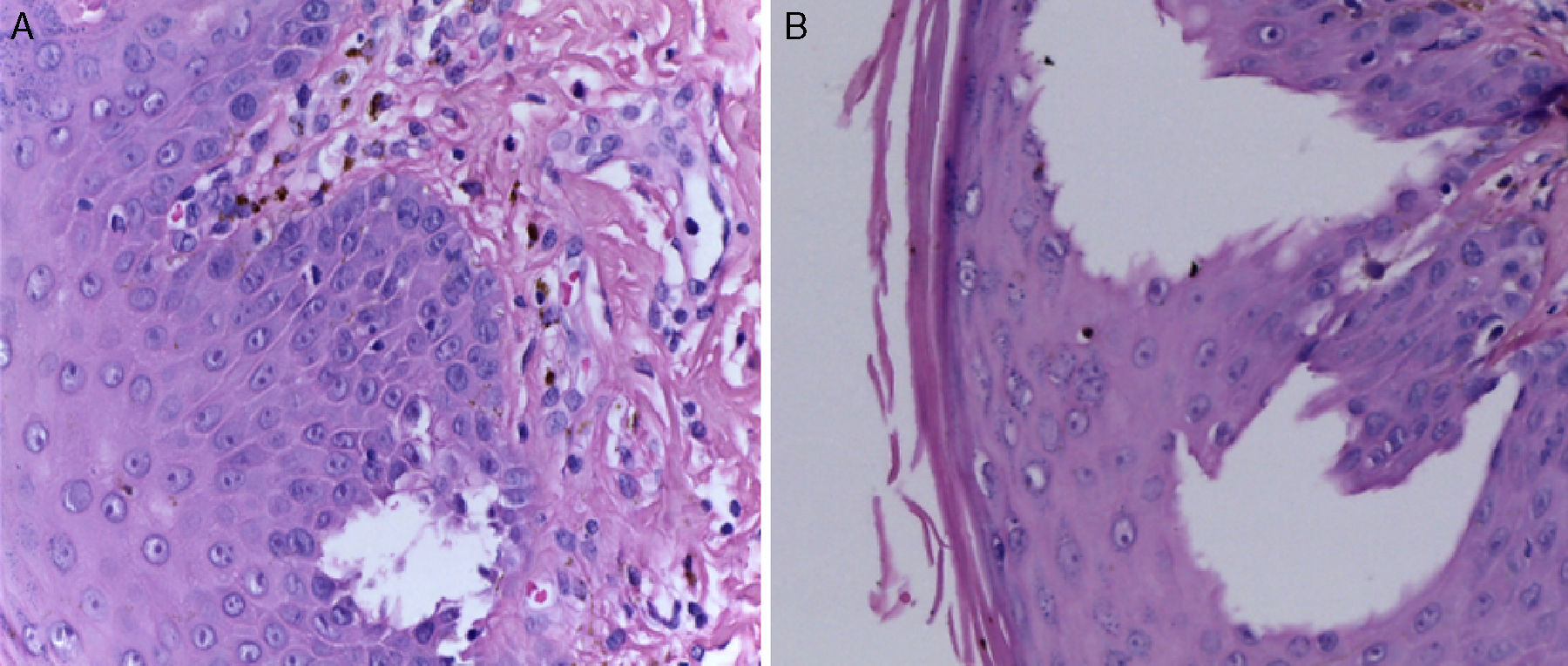
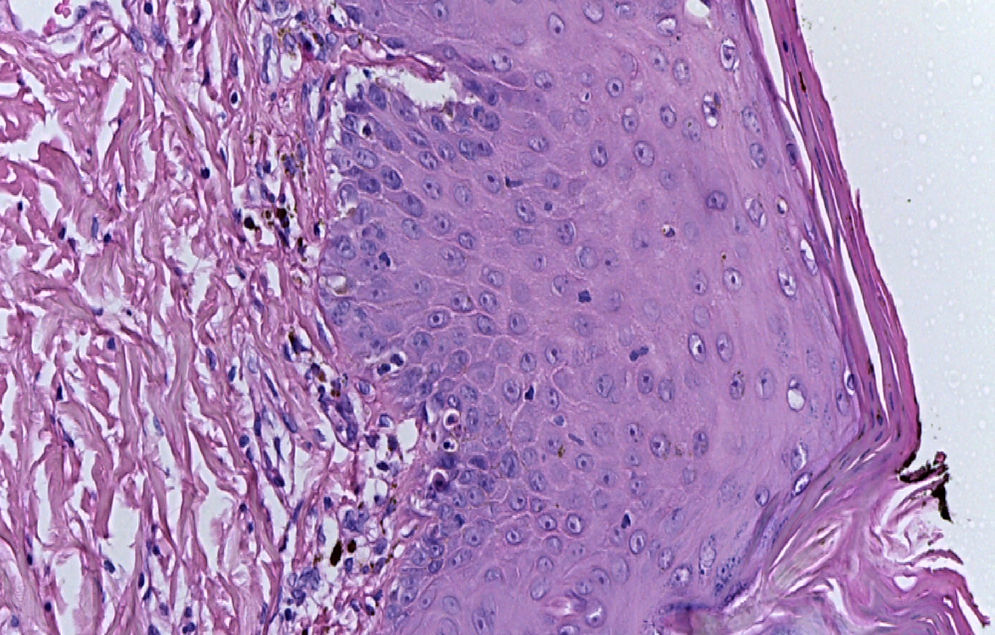
La biopsia de piel reportó piel con hiperqueratosis y paraqueratosis focal. El resto de la epidermis mostró disqueratosis, exocitosis de linfocitos y neutrófilos y formación de vesículas intraepidérmicas (fig. 4). En la unión dermoepidérmica y dermis papilar se observó infiltrado en banda constituido por linfocitos y melanófagos con pigmento (fig. 5). La dermis reticular y el tejido subcutáneo no mostraron alteraciones.
 Vesícula intraepidérmica y salida de pigmento a la dermis papilar. B) Vesícula intraepidérmica.")

Diagnóstico final
Incontienencia pigmenti fase III.
Posteriormente, la paciente tuvo una evolución tórpida. Persistió con inestabilidad hemodinámica y respiratoria, persistencia de las crisis convulsivas, sepsis y choque séptico refractario a aminas. El esquema antibiótico se progresó a meropenem, vancomicina y anfotericina-B. Sin embargo, a pesar de los cuidados intensivos y de la progresión oportuna del esquema antimicrobiano, desarrolló síndrome de falla orgánica múltiple con insuficiencia renal, insuficiencia hepática aguda, síndrome compartamental abdominal, síndrome de dificultad respiratoria aguda (SDRA) y coagulación intravascular diseminada (CID), persistió con hipotensión refractaria y acidosis metabólica, llenado capilar prolongado, pulsos débiles y bradicardia, lo que le condujo al paro cardiocirculatorio irreversible.
3DiscusiónEl presente caso corresponde a un lactante femenino con un cuadro clínico compatible con incontinencia pigmenti grave, que inició desde el periodo neonatal con afección cutánea, neurológica e inmunológica. Se pudieron descartar otras causas de eritema vesículo-ampolloso neonatal, como las infecciones por virus herpes simple y sífilis congénita, así como otras alteraciones de la pigmentación cutánea como la hiperpigmentación secundaria a la hiperplasia suprarrenal congénita —causada por la reactividad cruzada de la ACTH con los receptores para la hormona estimulante de los melanocitos (MSH)—y la hiperpigmentación transitoria benigna donde no existe afección de los anexos de la piel. El diagnóstico de incontinencia pigmenti se confirmó con el estudio histopatológico de la biopsia de piel.
Esta paciente presentó afección neurológica grave, relacionada con anomalía del desarrollo cerebral y trastornos en la migración neuronal con hipoplasia del cuerpo calloso, así como fenómenos vasculares con isquemia y hemorragia. Esto se desarrolló tempranamente, con crisis convulsivas de difícil control acompañadas de deterioro neurológico progresivo, lo cual complicó su condición clínica. Sin embargo, se debe destacar que a pesar de que en otros casos de incontinencia pigmenti se ha reportado la frecuente asociación de las alteraciones neurológicas con afecciones oftalmológicas, en esta paciente la única alteración ocular detectada fue el leucoma corneal.
Se consideró que la hipertensión arterial pulmonar manifestada por el aumento de la presión sistólica del ventrículo derecho (PSVD) se debía a la grave afección pulmonar secundaria al proceso neumónico que estaba cursando.
Sin embargo, este caso permite ilustrar cómo el diagnóstico de esta enfermedad se puede sospechar en los lactantes femeninos que acuden a los hospitales pediátricos en malas condiciones generales con sepsis y estado de choque, y que presenten por lo menos uno de los criterios diagnósticos mayores para la enfermedad, como exantema vesicular, hiperpigmentación cutánea, líneas de Blaschko, placas verrugosas y alopecia frontal, así como la presencia de alguno de los criterios diagnósticos menores propuestos por Landy y Donnai12.
En esta paciente no se pudieron realizar estudios moleculares para la determinación del tipo de mutación del gen NEMO ni pruebas para detectar un posible retraso en la inactivación del cromosoma X que porta la mutación debido a las condiciones clínicas de gravedad con las que se recibió. Tampoco se pudieron efectuar pruebas sobre la integridad y funcionamiento del eje de la interleucina-12. No obstante, la citometría de flujo para conteo de subpoblaciones de linfocitos mostró linfopenia grave (CD45 = 433, de lo cuales el 99% fueron linfocitos T (CD3 = 429) y solo el 1% de linfocitos B), con inmunoglobulinas en los límites normales bajos. Esto determinó el desarrollo de varias infecciones graves en su corta vida, así como su defunción causada por un evento de sepsis refractaria a pesar del tratamiento antibiótico adecuado. Lo anterior permite inferir que la paciente cursó con una alteración grave tanto de la inmunidad celular como de la inmunidad humoral.
Partiendo del hecho de que los tratamientos propuestos para esta rara enfermedad (gammaglobulina intravenosa y de gamma-interferón) son costosos y poco accesibles en nuestro medio, se considera que la posibilidad de ofrecer una terapéutica oportuna a las pacientes con incontinencia pigmenti dependerá del establecimiento precoz de un diagnóstico confirmado de la enfermedad. Esto además del soporte vital en unidades de cuidados intensivos de las enfermas graves e inestables, así como del abordaje y tratamiento multidisciplinario de las alteraciones infecciosas, neurológicas y oftalmológicas40–46. Finalmente, no debe olvidarse que es esencial proporcionar a los padres asesoramiento genético47–50.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.