


Shenqu is a fermented product that is widely used in traditional Chinese medicine (TCM) to treat indigestion; however, the microbial strains in the fermentation process are still unknown. The aim of this study was to investigate microbial diversity in Shenqu using different fermentation time periods. DGGE (polymerase chain reaction-denaturing gradient gel electrophoresis) profiles indicated that a strain of Pediococcus acidilactici (band 9) is the predominant bacteria during fermentation and that the predominant fungi were uncultured Rhizopus, Aspergillus oryzae, and Rhizopus oryzae. In addition, pathogenic bacteria, such as Enterobacter cloacae, Klebsiella oxytoca, Erwinia billingiae, and Pantoea vagan were detected in Shenqu. DGGE analysis showed that bacterial and fungal diversity declined over the course of fermentation. This determination of the predominant bacterial and fungal strains responsible for fermentation may contribute to further Shenqu research, such as optimization of the fermentation process.
Shenqu, also known as Liushenqu, is commonly used in Chinese medicine clinics to protect the stomach and spleen and stimulates appetite and digestion. Current research efforts have revealed that some digestive enzymes (amylase enzymes, protease enzymes, glucoamylase), vitamins and other substances play a main role in stimulating appetite and digestion.1 Resistance theory is the earliest work to mention Shenqu. Shenqu is traditionally processed as follows: wheat bran, flour, ricebean powder (Vigna umbellata [Thunb.] Ohwi and Ohashi), and bitter apricot seed powder (Prunus mandshurica [Maxim.] Koehne) are blended in a particular ratio. Various Chinese medicine decoctions are then added, including Polygonum pubescens (Blume), Xanthium sibiricum (Patr.), and Artemisia annua (L.). The mixture is then kneaded and divided into bricks, which are put into a mold. Finally, the bricks are covered with adhesive-bonded cloth and placed in a box at constant temperature and humidity. After a few days of fermentation, the product is cut into small lumps and dried at a low temperature. The quality of the resulting Shenqu can vary due to differences in the amount of the mixed bacteria and fungi that are present during fermentation. It is worth noting that the fungus Aspergillus flavus produces aflatoxin, a carcinogen, during fermentation. This is one of the reasons why Shenqu is not included in the Chinese Pharmacopeia. However, the current theoretical support endorses Shenqu for stimulating appetite and digestion.a better understanding of the microbes involved in Shenqu fermentation may lead to improved methods of fermentation.
There are two main types of methods for assessing bacterial diversity, traditional culture-dependent methods and culture-independent methods. Thus far, studies on the microbial diversity of Shenqu have been mainly based on traditional culture-dependent methods,2–4 such as PCR-SSCP (single strand conformation polymorphism)5 and DGGE.6 PCR-DGGE (polymerase chain reaction-denaturing gradient gel electrophoresis) is a culture-independent method designed to analyze the genetic diversity in a sample. It overcomes the disadvantages of culture-dependent methods,7 making it a common tool for molecular biological investigations into microbial communities. PCR-DGGE has been used widely to analyze microbial community structure across different fields, such as food microbiology, oral microbiology, soil microorganisms, environmental microbiology, and other areas.8–11 In this study, we used culture-independent PCR-DGGE and TA cloning to determine the microbial diversity of Shenqu across different fermentation periods. The aim of this study was to investigate eubacteria microbial diversity during fermentation and identify several dominant fermentation bacteria and fungus.
Materials and methodsShenqu sample collectionShenqu fermentation parameters were based on our previous study and response surface methodology.12 Raw materials were crushed in a grinder. Fourteen grams of Polygonum pubescens (Blume), Xanthium sibiricum (Patr.) and Artemisia annua (L.) were mixed with water and decocted for 1h at 32°C and 75% relative humidity and then mixed with 60g of flour, 140g of wheat bran, 8g of bitter apricot, and 5.2g of ricebean. Eight samples were processed and designated as 1–8 for fermentation for varying lengths of time, representing days 1–8, respectively. Each Shenqu sample, of approximately 100g, was collected during days 1–8. All samples were collected in a sterile environment, transferred to sterile polyethylene bags and stored at −70°C until they were analyzed.
DNA extractionFive grams of each Shenqu sample were suspended in 50mL of phosphate buffered saline (PBS, 0.1mol/L, pH 8.0) and shaken for 10min. The mixed suspension was centrifuged at 10,000×g for 10min and washed three times using the same PBS buffer. Total genomic DNA was extracted from the pellets using a ONE-4-ALL Genomic DNA Mini-Preps Kit (Sangon Biotech, Shanghai, China) according to the manufacturer's instructions. The samples were ground using liquid nitrogen and lysis buffer, then rapidly thawed in a water-bath at 65°C for an hour. The samples were shaken every 10min during lysis. The crude DNA was electrophoretically analyzed on 1.2% (w/v) agarose gels; samples were then kept in a clean 0.5-mL microcentrifuge tube and stored at −20°C.
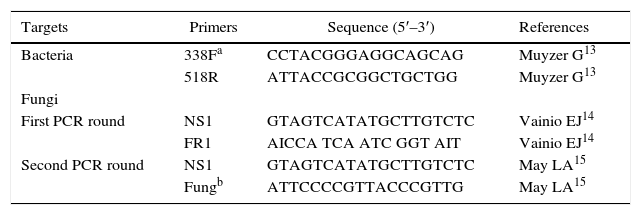
PCR amplificationAll primers used in this study are listed in Table 1. General bacterial 16S rRNA gene primers 338F and 518R were used to assess bacterial diversity. A touch-down PCR technique was employed in order to increase sensitivity. The thermal cycling conditions were as follows: 5min denaturation at 95°C; 5 cycles of 30s at 94°C, 30s at 62°C (with each cycle reduced by 2°C), and 90s at 72°C; 25 cycles of 30s at 94°C, 30s at 50°C, and 90s at 72°C; and final extension for 10min at 72°C. A GC clamp (5′-CGC CCG CCG CGC GCG GCG GGCGGG GCG GGG GCA CGG GGG G-3′) was attached to the 5′ end of primer 338F for the DGGE analysis.
Primers used in this study.
| Targets | Primers | Sequence (5′–3′) | References |
|---|---|---|---|
| Bacteria | 338Fa | CCTACGGGAGGCAGCAG | Muyzer G13 |
| 518R | ATTACCGCGGCTGCTGG | Muyzer G13 | |
| Fungi | |||
| First PCR round | NS1 | GTAGTCATATGCTTGTCTC | Vainio EJ14 |
| FR1 | AICCA TCA ATC GGT AIT | Vainio EJ14 | |
| Second PCR round | NS1 | GTAGTCATATGCTTGTCTC | May LA15 |
| Fungb | ATTCCCCGTTACCCGTTG | May LA15 | |
F, forward primer; R, reverse primer.
A nested PCR technique was employed in order to increase sensitivity. PCR amplification of general bacterial 18S rRNA was performed using universal gene primers NS1 and FR1 in the first step, followed by nested PCR using NS1 and GC-Fung. The thermal cycling conditions were as follows: 5min denaturation at 95°C; 30 cycles of 30s at 94°C, 30s at 50°C, and 90s at 72°C; and final extension for 10min at 72°C. PCR products from the first step were diluted with 10 times the amount of ddH2O and served as the template for the second round of nested PCR.
DGGE analysisThe PCR products of bacteria and fungi were analyzed using DGGE and the D-code Universal Mutation Detection System (Bio-rad, USA). For assessing bacterial diversity, 10% of the polyacrylamide gradient (acrylamide:bisacrylamide, 37.5:1) was used. The optimal separation was achieved by a 40–70% denaturant gradient. For assessing fungal diversity, 8% polyacrylamide and 25–40% denaturant gradient were used. Electrophoresis was then performed for 1h at 60V and 15h at 100V (60°C). After electrophoresis, gels were stained with SYBR Green I (Molecular Probes, BBI, Candia) for 30min. The gels were observed, and photographs were taken using a KETA G series Image System (Wletch, USA).
Sequencing of DGGE bandsRepresentative bands were excised from gels with a sterile blade. The gel pieces were ground using tissue-grinding pestles (Sangon, Shanghai, China) and then incubated overnight at 4°C in TE buffer (pH 8.0). The DNA solution with TE was then amplified with primers with no GC clamp. Purified PCR products were ligated into a pUCm-T vector and then transformed into Trans5α Chemically Competent Cells (Transgen Biotech, Beijing, China). Individual white colonies were amplified with PCR using the primers M13-4716 and M13-48 (Sangon, Shanghai, China). Samples were then sent to a sequencing company for sequencing (Sangon, Shanghai, China). The resulting gene sequences were aligned with those in a Gen Bank with the Blast program to identify the closest known relatives.
Statistical analysesQuantity One software (Bio-rad, USA) was used to analyze the DGGE profiles and perform cluster analysis. Statistical analysis of the data sets was performed using MATLAB 2013a software (Mathworks, USA). The Shannon–Wiener index was determined by the relative intensity of bands.
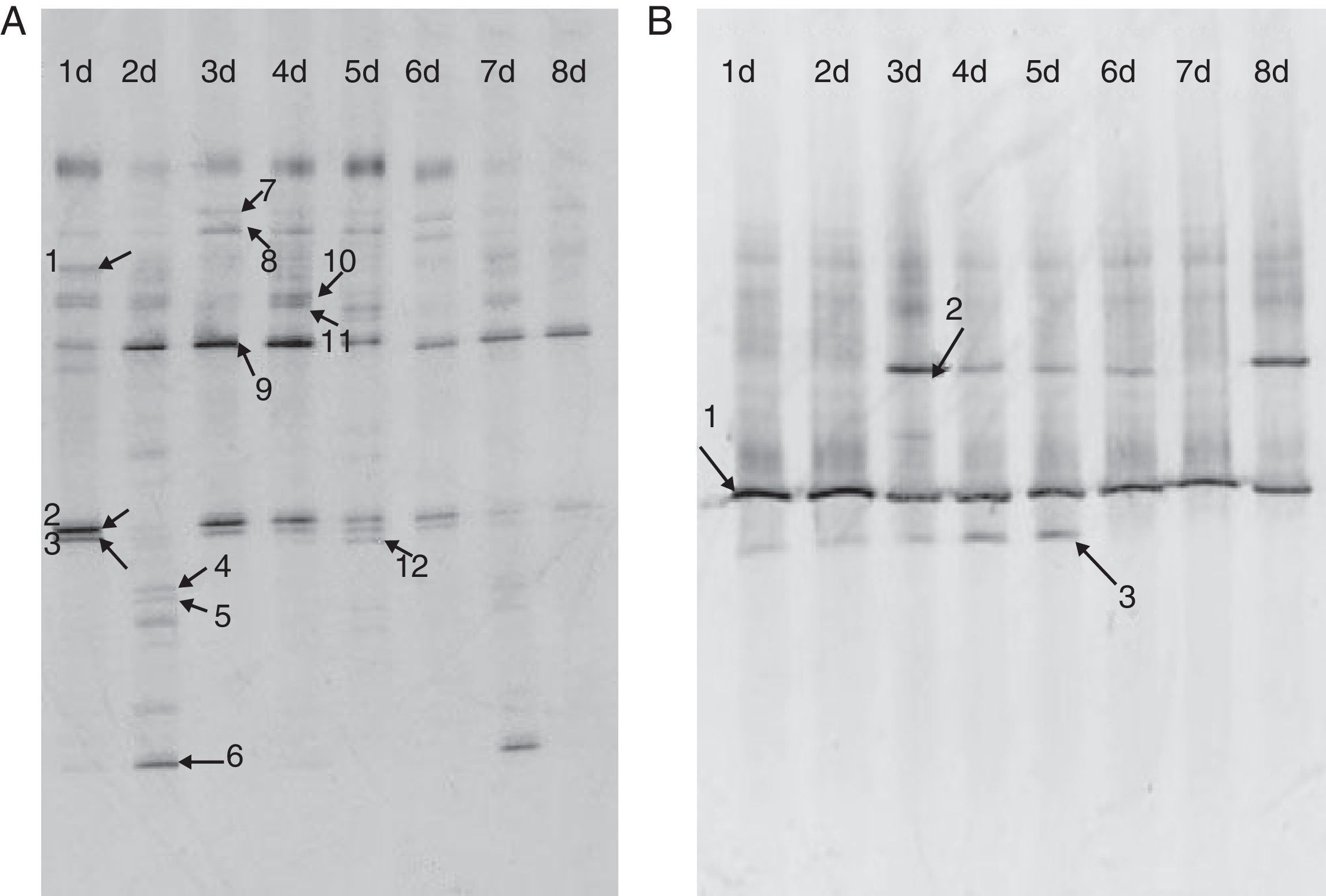
ResultsBacterial and fungal community diversityThe DGGE profile for the bacterial community of fermenting Shenqu is shown in Fig. 1. Notably, the bacterial community differed over the course of fermentation, while the fungal community did not differ. Diversity indices of microbes in Shenqu were calculated based on the DGGE profile. The bacterial diversity indices over 8 days of fermentation were as follows: day 1, 21 bands, Shannon–Wiener index 2.38; day 2, 23 bands, index 2.56; day 3, 13 bands, index 2.07; day 4, 13 bands, index 2.05; day 5, 18 bands, index 2.19; day 6, 18 bands, index 2.15; day 7, 19 bands, index 2.35; and day 8, 7 bands, index 1.52. The fungal diversity indices over the 8 days were as follows: day 1, 8 bands, Shannon–Wiener index 1.69; day 2, 10 bands, Shannon–Wiener index 1.92; day 3, 4 bands, Shannon–Wiener index 1.36; day 4, 8 bands, Shannon–Wiener index 1.77; day 5, 7 bands, Shannon–Wiener index 1.77; day 6, 7 bands, Shannon–Wiener index 1.35; day 7, 7 bands, Shannon–Wiener index 1.59; and day 8, 7 bands, Shannon–Wiener index 1.71. The species richness varied over the eight samples, and most bands were observed in the sample from day 2 (Fig. 1A and B). The sample from day 2 also had the highest Shannon–Wiener indices (2.56 and 1.92) of the PCR-DGGE profiles.
 A 40–70% denaturing gradient was used. (B) A 25–40% denaturing gradient was used.")
Touchdown PCR-DGGE and nested PCR-DGGE profile of bacterial community diversity of Shenqu from the 16s rDNA and 18s rDNA obtained from Shenqu after varying durations of fermentation. Lanes 1–8d refer to samples derived from the 1st to the 8th day of fermentation, respectively. (A) A 40–70% denaturing gradient was used. (B) A 25–40% denaturing gradient was used.
The sequencing of bacterial DGGE bands highlighted the presence of various bacterial strains, including Enterobacter cloacae (band 1, 100% identity to NCBI accession KM408606), Klebsiella oxytoca (bands 2 and 10, 100% identity to KM408607 and KM408615), Erwinia billingiae (bands 3 and 11, 100% identity to KM408608 and KM408615), Escherichia hermannii (band 4, 99% identity to KM408609), Paenibacillus polymyxa (band 5, 99% identity to KM408610), Pantoea vagans (band 6, 100% identity to KM408611), Acinetobacter baumannii (band 7, 100% identity to KM408612), Desulfotomaculum thermocisternum (band 8, 100% identity to KM408613), P. acidilactici (band 9, 99% identity to KM408614), and Citrobacter koseri (band 12, 100% identity to KM408617) (Fig. 1A). Notably, P. acidilactici (band 9, 100% identity to KM408614)) was detected throughout the entire fermentation process.
The sequencing of fungal DGGE bands highlighted the presence of three strains: uncultured Rhizopus (band 1, 100% identity to NCBI accession KM408618), Aspergillus oryzae (band 2, 100% identity to KM408619), and Rhizopus oryzae (band 3, 100% identity to KM408620) (Fig. 1B). Again, one species, the uncultured Rhizopus (band 1), was detected throughout the entire fermentation process, followed by band 2,3 (A. oryzae, R. oryzae).
DiscussionIn this study, PCR-DGGE was applied to analyze the microbial community structure of the TCM supplement Shenqu. Shenqu is a natural culture medium containing various nutrients. Conventional culture methods are unable to reflect its full nutritional contents. Therefore, our study adopted the culture-independent method of PCR-DGGE to investigate the bacterial and fungal community structure of Shenqu. The bacterial DGGE fingerprints showed that the Pediococcus acidilactici strain (band 9, Fig. 1A) was the predominant bacterial species present during fermentation. Likewise, the predominant fungus during fermentation was uncultured Rhizopus, followed by A. oryzae, and R. oryzae. From Berger's bacterial identification manual and related literature,17–19 we know that these bacteria can produce amylase, protease enzymes such as glucoamylase, and digestive enzymes. These products are likely to be associated with the appetite stimulating and digestive functions of Shenqu.
The sequencing results showed that the bacterial community included 10 types of pathogenic bacteria, including seven E. cloacae strains, K. oxytoca,20E. billingiae, and P. vagan.21 This study confirmed that pathogenic bacteria exist in the traditional Chinese medicine Shenqu. The existence of pathogenic bacteria is likely to affect the quality of various batches of Shenqu compared with batches of Shenqu that have undergone pure bred fermentation22,6 also investigated the microbial community of Shenqu by PCR-DGGE and found that the dominant microbes belonged to the genera Enterobacter, Pediococcus, Pseudomonas, Mucor, and Saccharomyces, which are results that are somewhat different from ours. This outcome is probably due to the different proportions of ingredients and fermentation parameters used in the two studies.
In conclusion, the aim of this study was to investigate the microbes of Shenqu over varying durations of fermentation by PCR-DGGE. The results revealed that P. acidilactici, A. oryzae, and R. oryzae were the predominant microbes present. These results may contribute to further study of Shenqu, such as studies focusing on optimizing the fermentation process or pure bred fermentation of Shenqu. Only by purifying the predominant microbes of Shenqu will we be able to examine the microbial biological transformations that occur in Shenqu. Thus, in this study, we suggest that PCR-DGGE should be considered as a preliminary tool for investigating the microbial community structure of Shenqu. Because of technical deficiencies of the PCR-DGGE method, however, some elements of the microbial community may inevitably go undetected. Other new technologies, such as T-RLFP, MLST and high-throughput sequencing, could therefore be adopted for further studies.
Conflicts of interestThe authors declare no conflicts of interest.
This work was financially supported by the National Science and Technology Major Projects Construction of the Incubator (Benxi) Base of National Innovation Drugs in Liaoning Province (20102X09401-304-105A).