





The study of metabolomics requires extracting as many metabolites as possible from a biological sample. This study aimed to determine the optimal method for the extraction of metabolites from solid-state fermented cottonseed meal (FCSM). The UPLC-Q-TOF-MS global metabolomics technology was used to detect the metabolites in FCSM, and the extraction quantity and extraction efficiency of seven different extraction methods, specifically the WA, 50MeOH, 50MeOHB, 50MeCNB, 80MeOHB, 80MeOH and AMF methods were evaluated. The results showed that the number of VIP metabolites extracted by AMF method are 196 and 184 in ESI+ and ESI− mode respectively, it is the largest number of all exacted methods; and the AMF methods also provided a higher extraction efficiency compared with the other methods, especially in indoleacrylic acid, dl-tryptophan and epicatechin (p<0.01). As a result, AMF/−4°C method was identified as the best method for the extraction of metabolites from FCSM by Lactobacillus acidophilus. Our study establishes a technical basis for future metabolomics research of fermented feed.
Cottonseed meal (CSM) is the most important plant protein in the world. However, its application in animal husbandry is limited due to its complex content, which includes free gossypol (FG), cyclopropene fatty acids (CPFA) and other anti-nutritional factors, such as a high fibre content, poor protein profile, etc. The most critical problem in CSM is FG, which has a negative effect on animal production.1–3 Microbial fermentation was used to promote the application and popularization of CSM because it could reduce the FG effectively and improve the nutritional value of CSM, as reported recently.4–7Lactobacillus acidophilus is an important lactic acid bacteria and intestinal microbe that regulates the intestinal micro-flora balance, enhances immunity, and reduces cholesterol levels.8–10 But its use for feed fermentation remains rare and few studies have investigated solid-state fermented cottonseed meal (FCSM) by L. acidophilus.
A prerequisite for a metabolomics study of the target metabolites of FCSM by L. acidophilus is the optimization of the pretreatment of the biological samples, including scientific sampling, reasonable quenching and high-efficiency extraction.11,12 The ideal method should produce a sample of all intracellular and extracellular metabolites simultaneously for the analysis of complex biological samples for global metabolomics.13 Both intracellular and extracellular metabolites exert a significant effect on the results of microbial metabolomic studies.14 Fermented feed is a complex biological sample; the fermentation products not only include the metabolites of microbial fermentation but also include the fermentation substrate nutrients. To truly reflect the fermentation process, the pretreatment methods of quenching and extracting metabolites must be optimized.
Previous studies of the pretreatment of biological samples have demonstrated that different quenching and extracting methods were suitable for different microorganisms, such as Escherichia coli, Yeast, Lactic acid bacteria, etc.15–19 These studies mainly included extracting microorganism metabolites by screening different pretreatment methods and examining the effects of different quenching solvents on the extraction of metabolites and sample grinding degree. However, these evaluations studied how to use different research platforms and indicators of the extraction method. The majority of studies were of the extraction of different quenching solvents, with a research platform based on traditional GC–MS and LC–MS. Recent developments have proved that UPLC-Q-TOF-MS could detect complex multi-component substances more effectively, faster and more accurately than other methods.20–22 At the same time, the UPLC-Q-TOF-MS platform can also be analysed quantitatively for the number of metabolites. So far, a few applications of L. acidophilus to metabolomics have been studied. Since L. acidophilus fermented protein feed is rare for traditional feed production, the method of extracting metabolites from FCSM has not received sufficient attention. Therefore, it was desirable to establish an extraction method that can be applied to a wide range of metabolites from L. acidophilus.
To identify the final metabolites in FCSM by L. acidophilus, both intracellular and extracellular metabolites must be analysed. We used the global metabolomics profiling way to evaluate the efficiency of seven extraction methods (WA, 50MeOH, 50MeOHB, 50MeCNB, 80MeOHB, 80MeOH and AMF) systematically with the aim of identifying the best method for the extraction from the FCSM. Seven different pretreatment methods were evaluated through a global metabolomics analysis based on UPLC-Q-TOF-MS. The quantity and efficiency of the metabolites extracted from FCSM were evaluated. This study will provide a good basis for future analyses of the total metabolites in the FCSM.
Materials and methodsChemicalsLC–MS-grade methanol and acetonitrile were purchased from Fisher Chemical (Fisher Scientific, USA) or Sigma–Aldrich (St. Louis, MO, USA). The water was purified with an EPED-E2-T device (Nanjing, China).
Sample preparationL. acidophilus (JJ-0787) was acquired from the China Center of Industrial Culture Collection (CICC). Fermentation substrates were composed of 65.89% CSM and 34.11% corn, with a total protein of 30%. JJ-0787 strains were cultured at 37°C for 24h in MRS broth (Solarbio Life Sciences Ltd., Beijing, China), and then inoculated 6mL JJ-0787 strains culture fluid in 100g sterilized fermentation substrate (the moisture content is 40%), mixed thoroughly, sealed in the triangle bottle, and then statically placed in an incubator at 37°C for 48h. Each group contained three repetitions. The resulting samples of FCSM were immediately frozen in liquid nitrogen and then stored at −80°C until analysis.
Metabolite extractionTo investigate the optimal method for the extraction of the metabolites in FCSM, the different quenching solvents and pretreatment methods used in the studies conducted by Tokuoka and Chen were modified as follows.18,19
WA (J1 group): A sample of FCSM was treated with high-speed homogenization (HHM). Then, a 0.2-g sample was rapidly quenched and dissolved in 2mL of ultra-pure water (4°C). The samples were quickly homogenized for 10min using a high-flux tissue homogenizer (TisssueLyser II, Qiagen, Germany). The centrifuge tubes were incubated for 30min on ice. The extract solution was centrifuged (−20°C, 10,000r/min, 5min), and the residual debris was removed. The supernatant was dissolved in 10 volumes of ultra pure water (4°C) and filtered with a 0.22-μm syringe filter (004022NL-SEPTFE, Shanghai, China). The samples were stored at −80°C until further analysis.
50MeOH (J2 group): The extraction process was the same as that used in the WA method except that the quenching solvent was replaced by a 50% cold methanol solution (methanol:water=50:50, −20°C).
50MeOHB (J3 group): The extraction process was identical to that used in 50MeOH method except that the 50% cold methanol solution (−20°C) was replaced with a 50% cold methanol solution (room temperature), and after the addition of quenching solvent to the sample, the method incorporated a boiling procedure (70°C, 5min) immediately followed by the same process as used in 50MeOH method.
50MeCNB (J4 group): The extraction procedures were identical to those used in 50MeOHB method with the exception that the 50% methanol solution (room temperature) was replaced by a 50% acetonitrile solution (acetonitrile:water=50:50, room temperature).
80MeOHB (J5 group): The 50% methanol solution used in 50MeOHB method was replaced with a 80% methanol solution (methanol:water=80:20, room temperature), followed by the same method as in 50MeOHB.
80MeOH (J6 Group): The extraction procedures were identical to those used in 50MeOH method except that the 50% cold methanol solution was replaced with an 80% methanol solution (methanol:water=80:20, −20°C), followed by the same method as in 50MeOH.
AMF (J7 group): The 50% cold methanol solution used in the 50MeOH method was replaced by a cold AMF solution (acetonitrile:methanol:water=2:2:1, formic acid 0.1mol/L, −4°C), and the other methodological details are the same as those used in the 50MeOH method.
The samples were diluted 20-fold with the corresponding extraction solvent, vortexed and centrifuged at 13,200r/min for 2min. A 150μL volume of supernatant from each sample was analysed by UPLC-Q-TOF-MS. Finally, the metabolites were identified and analysed based on the obtained data.
UPLC-Q-TOF-MS detection methodThe chromatographic conditions were as follows: The analysis was performed on an ACQUITY UPLC BEH, which was configured with direct injection using a C18 column (100m, 2.1mm, 1.7mm, Waters, USA). The column volume was 10μL, and the column temperature was set to 35°C. The mobile phase A was ultra pure water, while mobile phase B was HPLC-grade acetonitrile with 0.1% (v/v) formic acid, and the flow rate was 0.35mL/min starting with 2% acetonitrile up to the full gradient of 98% in mobile phase B (0–12min). After equilibration for 1min, mobile phase B ranged from 98% to 2% for 1min and equilibration for 1min after the sample was collected. The injection volume was 2μL, the column temperature was set to 35°C, and the autosampler temperature was maintained at 4°C.
Mass spectrometry conditions: An electrospray ionization (ESI) source was used to scan in the positive and negative ionization modes. The ion source temperature was 120°C, and the desolvation temperature was 500°C. The nitrogen flow rate for solvent evaporation was 800L/h, and the flow rate of tapered blowing nitrogen was 50L/h. The positive- and negative-ion mode capillary ionization voltages were 3.0 and 2.5kV, respectively; the sampling cone voltages were 27 and 30V, respectively; the collision energy was 6eV; and the sampling cone voltages were 27 and 30V, respectively. The collision energy was 6eV, and the quadrupole scanning range was between 50 and 1000m/z.
Data statistics and analysisThe raw data detected by UPLC-Q-TOF-MS were analysed by XCMS software (http://xcmsonline.scripps.edu) for peak identification and matching.23 The chromatographic peak data were normalized uniformly, and the multidimensional data were analysed using SIMCA-P software (version 13.0, Sweden). Data pre-processing was performed using the Pareto Scaling method. A partial least squares-discriminate analysis (PLS-DA) was then used to perform supervised multidimensional statistical analysis, and the permutation test was used to prevent over-fitting of the PLS-DA model.24 The differentiated expressed metabolites (VIP metabolites) were identified and the number of different metabolites were compared through a t-test (p value <0.05) and fold-changes (fold change >2 or <0.5), revealing a multiple relationship between the two groups.22
Tandem mass spectrometry was used to identify metabolites based on exact molecular weights (molecular weight error of <30ppm). The Masslynx i-FIT algorithm was used to further confirm the possible structural formulas according to the isotope principle. Subsequently, accurate MS/MS's fragmentation information was queried in the Human Metabolome Database (HMDB, http://www.hmdb.cawebcite) and the Metlin database (http://metlin.scripps.edu/website) for the screening and identification of potential biomarker substances.25–27 The data were analysed by ANOVA using the SPSS22.0 statistical software. Multiple comparisons were performed using the Duncan method. The test data were expressed as the mean±SD, and p<0.05 was used as the criterion for significant differences.
ResultsPLS-DA analysisThe PLS-DA 3D scoring chart (Fig. 1) shows that the different metabolite extraction methods are clearly divided into three-dimensional spatial arrangements. The results showed significant differences in type, amount and concentration of metabolites extracted from different FCSM samples using the different pretreatment methods. All of the samples were within the 95% confidence interval and almost all of the groups were distinguishable from the others.
Analysis of the number of metabolites![PLS-DA 3D score plots for the extracted metabolites of different pretreatments. (A) PLS-DA 3D score plot in ESI+. (B) PLS-DA 3D score plot in ESI−. t[1]–t[3] indicating the separation between different groups.](https://static.elsevier.es/multimedia/15178382/0000004900000002/v2_202011260709/S1517838217303209/v2_202011260709/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNerEcKXq/4gYMpNkXqSlVDY9W8lBgxJd7sP21C796+Vu46WFcjsri9wYxk2OQ4nTh+uMlM+JS3YOSll4RM7HOOnK7Kzf50St0bCKhSxHg1PEQ/enwGNhrMfFBAVHrL2Huw5FxBNSGSyrU8mRWYunYTeWUWex7Lg53bc1nA8Y+CCD89/Y93xpDbkCULYgBN92PGoJ+wqP7SYjiFcKDM7SdcRDbtrXISZO063TAJnO37f2+f2gvks+Z6iQQYd1laAmME= "PLS-DA 3D score plots for the extracted metabolites of different pretreatments. (A) PLS-DA 3D score plot in ESI+. (B) PLS-DA 3D score plot in ESI−. t[1]–t[3] indicating the separation between different groups.")
The numbers of metabolites extracted using different methods are shown in Fig. 2. Preliminary evaluation of the merits of the extraction method depending on the amount of small-molecule metabolites detected by the different ion scan (ESI+ and ESI−) modes can allow a comprehensively characterization of the samples.
 Comparison of the number of metabolites after molecular feature extraction. Different lowercase letters means that there are significantly different (p<0.05) in each same serious bar. (B) Comparison of the number of VIP metabolites (fold change >2 or <0.5) between every experiment group and control group. J1 – WA, J2 – 50MeOH, J3 – 50MeOHB, J4 – MeCNB, J5 – 80MeOHB, J6 – 80MeOH, J7 – AMF.")
Comparison of the number of metabolites extracted with different pretreatment in ESI+ and ESI− mode. (A) Comparison of the number of metabolites after molecular feature extraction. Different lowercase letters means that there are significantly different (p<0.05) in each same serious bar. (B) Comparison of the number of VIP metabolites (fold change >2 or <0.5) between every experiment group and control group. J1 – WA, J2 – 50MeOH, J3 – 50MeOHB, J4 – MeCNB, J5 – 80MeOHB, J6 – 80MeOH, J7 – AMF.
As shown in Fig. 2A, an analysis of the metabolites revealed by scanning in the ESI+ mode showed that the J1 group had the lowest and the J6 group had the highest numbers of metabolites. The number of extracted metabolites in the J6, J2, J3 and J4 groups were increased 12.55, 11.70, 11.08 and 10.98%, respectively, compared with that in the J1 group (p<0.05). The numbers of metabolites in the J5 and J7 groups were higher than that in the J1 group, but the difference was not significant (p>0.05). The groups can be sorted as follows based on the number of metabolites, J6>J2>J3>J4>J5>J7>J1. After scanning in the ESI− mode, the numbers of metabolites in the J1group is the fewest in all groups, and the J2 group was the highest in all groups. The numbers of extracted metabolites in the J2, J5, J3 and J6 groups were increased by 20.91, 18.85, 17.86 and 17.15%, respectively, compared with the J1 group (p<0.05). The J4 and J7 groups had more metabolites than the J1 group, but the differences were not significant (p>0.05). The groups can be ordered based on the number of metabolites as follows: J2>J5>J3>J6>J4>J7>J1.
Fig. 2B shows the numbers of VIP metabolites in each group compared with the control group. After scanning in both the ESI+ and ESI− modes, the J7 group had more VIP metabolites than the other groups, and the J3 group had the fewest VIP metabolites. The groups can be ordered as follows based on the number of VIP metabolites identified in the EDI+ and ESI− modes was J7>J6>J4>J2>J5>J3 and J7>J6>J5>J4>J2>J3, respectively.
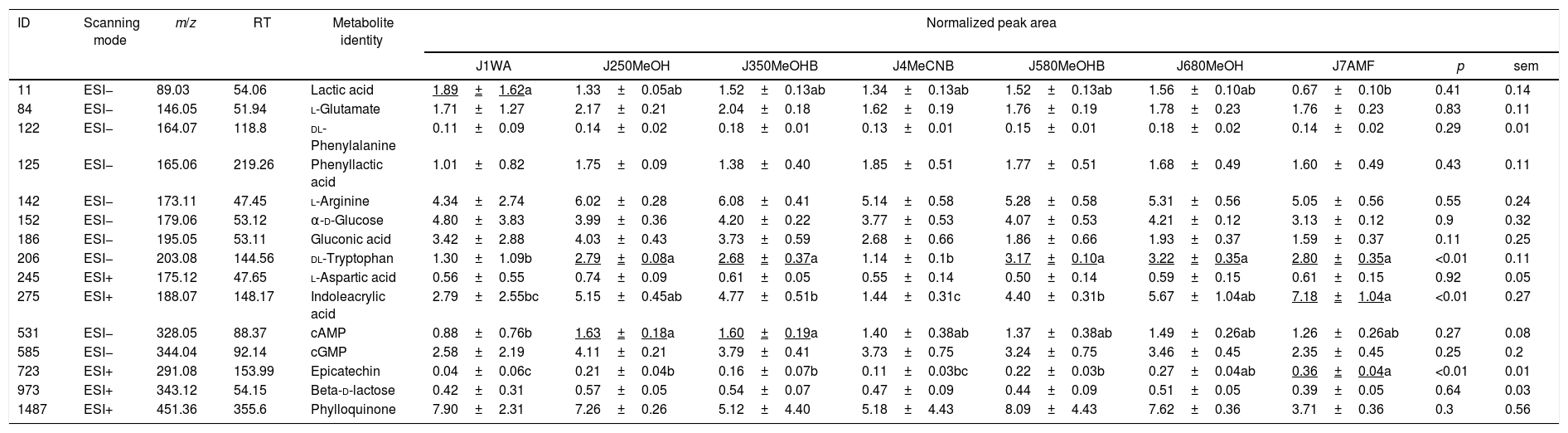
Comparison of extraction efficiency of common VIP metabolitesThe metabolite samples of different extraction methods were analysed by UPLC-Q-TOF-MS. Fifteen common peak variables were screened in the ESI+ and ESI− modes, and 15 kinds of small-molecule metabolites were identified by m/z and RT correlation databases, as shown in Table 1. Compared with the normalized peak area of the metabolites, the peak area values of dl-tryptophan, indoleacrylic and epicatechin in seven groups were very significantly different (p<0.01). The peak area value of dl-tryptophan was higher in the J6, J5, J7, J2, and J3 groups. The J6 group showed the highest value, but the difference between the five groups was not significant. There were significant differences between the five groups compared with those in groups J1 and J4 (p<0.01). The largest peak area for the indoleacrylic acid level was found for the J7 group, and this group revealed very significant (p<0.01) differences compared with the J1 and J4 groups, but the differences with the J6 and J2 groups were not significant (p>0.05). The highest peak area of epicatechin was found for the J7 group, but this area was not significantly (p>0.05) different compared with the J6 group. The J7 group presented significant differences compared with the J5, J2, J3, and J4 groups (p<0.05) and a very significant difference compared with the J1 group (p<0.01). In addition to the above-mentioned metabolites, there was a significant difference between cAMP and lactic acid. For the peak area of cAMP, the J2 and J3 group were higher than the J1 groups (p<0.05), but there was no significant difference with the other groups (p>0.05). For the peak area of lactic acid, the J1 group was the highest and there was a significant difference between the J7 and J1 groups (p<0.05). However, the other groups and the J1 group had no significant differences (p>0.05). There was no significant difference between the other metabolites (p>0.05) besides above metabolites.
Normalized peak area of metabolites extracted by different quenching method.
| ID | Scanning mode | m/z | RT | Metabolite identity | Normalized peak area | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| J1WA | J250MeOH | J350MeOHB | J4MeCNB | J580MeOHB | J680MeOH | J7AMF | p | sem | |||||
| 11 | ESI− | 89.03 | 54.06 | Lactic acid | 1.89±1.62a | 1.33±0.05ab | 1.52±0.13ab | 1.34±0.13ab | 1.52±0.13ab | 1.56±0.10ab | 0.67±0.10b | 0.41 | 0.14 |
| 84 | ESI− | 146.05 | 51.94 | l-Glutamate | 1.71±1.27 | 2.17±0.21 | 2.04±0.18 | 1.62±0.19 | 1.76±0.19 | 1.78±0.23 | 1.76±0.23 | 0.83 | 0.11 |
| 122 | ESI− | 164.07 | 118.8 | dl-Phenylalanine | 0.11±0.09 | 0.14±0.02 | 0.18±0.01 | 0.13±0.01 | 0.15±0.01 | 0.18±0.02 | 0.14±0.02 | 0.29 | 0.01 |
| 125 | ESI− | 165.06 | 219.26 | Phenyllactic acid | 1.01±0.82 | 1.75±0.09 | 1.38±0.40 | 1.85±0.51 | 1.77±0.51 | 1.68±0.49 | 1.60±0.49 | 0.43 | 0.11 |
| 142 | ESI− | 173.11 | 47.45 | l-Arginine | 4.34±2.74 | 6.02±0.28 | 6.08±0.41 | 5.14±0.58 | 5.28±0.58 | 5.31±0.56 | 5.05±0.56 | 0.55 | 0.24 |
| 152 | ESI− | 179.06 | 53.12 | α-d-Glucose | 4.80±3.83 | 3.99±0.36 | 4.20±0.22 | 3.77±0.53 | 4.07±0.53 | 4.21±0.12 | 3.13±0.12 | 0.9 | 0.32 |
| 186 | ESI− | 195.05 | 53.11 | Gluconic acid | 3.42±2.88 | 4.03±0.43 | 3.73±0.59 | 2.68±0.66 | 1.86±0.66 | 1.93±0.37 | 1.59±0.37 | 0.11 | 0.25 |
| 206 | ESI− | 203.08 | 144.56 | dl-Tryptophan | 1.30±1.09b | 2.79±0.08a | 2.68±0.37a | 1.14±0.1b | 3.17±0.10a | 3.22±0.35a | 2.80±0.35a | <0.01 | 0.11 |
| 245 | ESI+ | 175.12 | 47.65 | l-Aspartic acid | 0.56±0.55 | 0.74±0.09 | 0.61±0.05 | 0.55±0.14 | 0.50±0.14 | 0.59±0.15 | 0.61±0.15 | 0.92 | 0.05 |
| 275 | ESI+ | 188.07 | 148.17 | Indoleacrylic acid | 2.79±2.55bc | 5.15±0.45ab | 4.77±0.51b | 1.44±0.31c | 4.40±0.31b | 5.67±1.04ab | 7.18±1.04a | <0.01 | 0.27 |
| 531 | ESI− | 328.05 | 88.37 | cAMP | 0.88±0.76b | 1.63±0.18a | 1.60±0.19a | 1.40±0.38ab | 1.37±0.38ab | 1.49±0.26ab | 1.26±0.26ab | 0.27 | 0.08 |
| 585 | ESI− | 344.04 | 92.14 | cGMP | 2.58±2.19 | 4.11±0.21 | 3.79±0.41 | 3.73±0.75 | 3.24±0.75 | 3.46±0.45 | 2.35±0.45 | 0.25 | 0.2 |
| 723 | ESI+ | 291.08 | 153.99 | Epicatechin | 0.04±0.06c | 0.21±0.04b | 0.16±0.07b | 0.11±0.03bc | 0.22±0.03b | 0.27±0.04ab | 0.36±0.04a | <0.01 | 0.01 |
| 973 | ESI+ | 343.12 | 54.15 | Beta-d-lactose | 0.42±0.31 | 0.57±0.05 | 0.54±0.07 | 0.47±0.09 | 0.44±0.09 | 0.51±0.05 | 0.39±0.05 | 0.64 | 0.03 |
| 1487 | ESI+ | 451.36 | 355.6 | Phylloquinone | 7.90±2.31 | 7.26±0.26 | 5.12±4.40 | 5.18±4.43 | 8.09±4.43 | 7.62±0.36 | 3.71±0.36 | 0.3 | 0.56 |
RT, Retention Time; ESI, Electronic Spray Ion; cAMP, cyclic Adenosine monophosphate; cGMP, cyclic guanosine monophosphate.
Note: p<0.05 indicate that there is significantly difference in normalized peak area. p<0.01 indicate that there is very significantly difference in normalized peak area. Underlined number represent significant increased values (p<0.05).
The fold changes in peak area were calculated using the formula log2 for the ratio of normalized peak area values of the metabolites extracted by different methods to the control group. The fold change in the peak area reflected the degree of change in metabolite concentration between the trial group and the control group. As Fig. 3 shows, the comparison of ploidy area variation is seen in approximately 15 common small molecule metabolites from FCSM by different extraction methods. It can be seen that the change in the fold change area between J7 and J1 group was the most significant (p<0.05).

Peak areas fold change of 15 kinds of metabolites extracted from FCSM with difference pretreatments. Fold change >0 means that the relative content of metabolites is improved, while <0 means that it is reduced. J1 – WA, J2 – 50MeOH, J3 – 50MeOHB, J4 – MeCNB, J5 – 80MeOHB, J6 – 80MeOH, J7 – AMF.
Sample pretreatment is a key step in the study of biological system metabolites because it directly affects the extraction efficiency of intracellular and extracellular metabolites from fermented feed samples. So it is necessary to study the pretreatment methods used in metabolomics study of fermented feeds. Many pretreatment methods for the quenching and extracting of metabolites from biological samples have been proposed; however, since Koning and Dam proposed a rapid quenching method involving the directly addition 60% methanol at −40°C and followed by centrifugation of the cells,28 this technique has not been changed and remains the most common method for the sampling of microbial cultures.15,29,30 By comparing different quenching methods for E. coli, Winder also demonstrated that 60% cold methanol solution (−48°C) is the most suitable E. coli quench method and recommended multiple cycles of −48°C 100% methanol freeze–thaw extraction to extract metabolites effectively.17 However, the study found that different methanol/water ratios have different metabolic effects significantly, with the cold methanol/water extraction method extracting triphosphate compounds and other energetic compounds in E. coli metabolites (e.g., ATP, CTP, UTP, etc.) and that cold methanol/water extraction was not necessarily the best way to extract E. coli intracellular metabolites. Based on the study conducted by Kimball,31 Rabinowitz compared the effects of 10 quenching solvents on the extraction of E. coli metabolites on the basis of LC/MS analysis of the peak height of each peak compared to the total metabolite yield.32 The study found that the optimal extraction yield was obtained using AMF (acidic acetonitrile/methanol/aqueous solution). Tokuoka conducted a systematic study of 11 methods for the quenching of Aspergillus oryzae-fermented samples using 50MeCNB (50% acetonitrile/water) as the extraction solvent.18 The use of a water bath at 70°C water bath for 5min was identified as the most efficient extraction method. Methods involving quenching with 60% methanol/water, 80% methanol/water and 80% methanol/glycerol at −40°C were used to study the effect of 80% methanol/water on extraction. These solvents can effectively reduce the degree of metabolite leakage and intracellular metabolite levels more suitable for the quenching of Lactobacillus bulgaricus.19 Different samples and research objectives are associated with different extraction requirements, and the optimal extraction method will vary from that recommended by the above study. At present, there is no standard operating procedure (SOP) for the extraction of different biological samples. To extract the metabolites from different samples, the corresponding pretreatment method was used. It required optimization and standardization of the sample quenching and extraction method, which is the only way for the future development of metabolomics. Based on the results of the above studies, methanol, acetonitrile and acidic acetonitrile/methanol/water (AMF) were chosen as the three main quenching solvents for our study. We also set up two gradient (50%, 80%) quenching solvents to study. Simultaneously, the differences in extraction methods between cold methanol and hot methanol were investigated. The control group was selected by ultra-pure water extraction. Thus, in this experiment, we have chosen seven extracting methods for systematic assessment.
The development of LC/MS, GC/MS and other technology platforms allows for the simultaneous qualitative and quantitative determination of a large number of probiotic fermentation metabolites. In this study, UPLC-Q-TOF-MS was used as the research platform. UPLC and Time-of-Flight Mass Spectrometry (TOF-MS) can be used to perform multi-component analyses of complex materials. Ultra-high-performance liquid chromatography (UPLC) would be a great boost in the field of microbial fermentation metabolites research.20–22,33,34 The platform ensures the accuracy, high efficiency and reliability of the test results. In the present study, we found that 15 different small molecule metabolites can be extracted by seven different extraction methods through the global metabolomic analysis, including amino acids, organic acids, vitamins and small molecular active substances associated with the metabolites of beneficial microbial fermentation protein feeds. These metabolites play an important role in the growth, development and immunization of animals.
The number of extracted metabolites and the number of VIP metabolites were used as the preliminary evaluation indexes, especially the number of VIP metabolite, it can reflect the extraction accuracy for the metabolites. From the PLS-DA 3D score (Fig. 1), it was found that different quenching extraction methods had different extraction effects. The number of detectable peaks was also significantly different, which means that there was a clear distinction between the number of detectable metabolites and how much the number of VIP metabolites reflected the number of small molecule metabolites that may play a role in the detection of metabolites. In contrast to the number of detectable metabolites, it was clear from Fig. 2A that 80MeOH (J6) was optimal in the positive ion mode (ESI+) and 50MeOH (J2) was optimal in the negative ion mode (ESI−). The positive and negative ion mode metabolite sum ranking trend was 50MeOH (J2)>80MeOH (J6)>50MeOHB (J3)>80MeOHB (J5)>50MeCNB (J4)>AMF (J7)>WA (J1). However, in contrast to the number of VIP metabolites (Fig. 2B), we found that the ranking trend was AMF (J7)>80MeOH (J6)>80MeOHB (J5)>50MeCNB (J4)>50MeOH (J2)>50MeOHB (J3) in positive and negative modes. The AMF method (J7) has a significant advantage in either positive or negative ion modes. To compare the extraction efficiency of different methods of quench extraction, the fifteen common metabolites extracted from seven methods were screened and the normalized peak area and fold change index of these metabolites were compared (Table 1 and Fig. 3). The results showed that significantly different metabolites included indoleacrylic acid, dl-tryptophan and epicatechin. The extraction efficiency of these metabolites was the best in AMF (J7), followed by 80MeOH (J6) and 50MeOH (J2). It is worth mentioning that for 50MeOHB (J3), the peak area value of extracts of these metabolites was also higher, with an extraction efficiency second only to the above three methods.
The differences in the extraction efficiency of various methods may be closely related to the polarity, water content and temperature of the extraction solvent. All the reagents used in this experiment were analytical-grade organic solvents, in which the polarity of methanol is higher than that of acetonitrile. However, the water content of the extraction solvent will also affect the polarity. The content of solvent moisture in the AMF, 80MeOH and 80MeOHB solvents was 20%; the 50MeOH, 50MeOHB and 50MeCNB solvent moisture content was 50%; and the water content of the control group (J1) reached 100%. The study showed that the water content in the solvent directly affects the extraction efficiency, which were consistent with Kimball's report.31 Kimball proved that the high yield of nucleoside was related to the decomposition of nucleotides in the extraction solvent enriched in water. The different concentrations of cold methanol/water lead a huge deviation in the extracted metabolites. A higher the water content, a worse the extraction efficiency of metabolites. In this study, the extraction temperatures of the different extraction methods were also different, 50% acetonitrile at −4°C and a water bath at 70°C during the extraction. The 50% and 80% hot methanol solution were also heated in a water bath at 70°C, and the temperature of the ultra pure water was −4°C. Some studies have confirmed that low temperature extraction can extract metabolites more efficiently.15,17 Winder even proposed a method of multi-cycle freeze–thaw extraction of metabolites using −48°C 100% methanol. However, some studies have confirmed that heat extraction is also an effective method of extracting metabolites.17 For example, a method for quenching fermented samples of A. oryzae was studied using 50% acetonitrile/water (v:v=50:50) for 5min in a 70°C water bath,18 but the specific reason for the temperature difference in the extraction of metabolites remains to be elucidated.
The results showed that the quality performance of the seven quenching and extraction methods was AMF>80MeOH>50MeOH>80MeOHB>50MeOHB>50%MeCNB>WA. Therefore, we can see that the AMF method was the best method to extract FCSM with L. acidophilus. These test results were consistent with Rabinowitz.32 His experiments confirmed that the acidic acetonitrile/methanol/water solution not only reduced the degradation of triphosphate but also reduced the loss of high-energy metabolites and promoted its conversion to low-energy derivatives, which ultimately improved the extraction concentration of metabolites. It has a higher extraction efficiency, but Rabinowitz's research object was E. coli, and this study was of L. acidophilus FCSM, indicating that the method also has applicability.
ConclusionsIn conclusion, this study only focused on the comparison of metabolite extraction methods from FCSM with L. acidophilus for metabolomic protocols. The experimental results showed that the −4°CAMF solution extraction method was the most accurate representation and the highest extraction of the metabolites of FCSM by Lactobacillus. Since metabolites differ remarkably depending on microorganisms and growth conditions, a good extraction method might only work in certain situations. A standard extraction method was difficult to establish, and different extraction methods may need to be used for different study objects.
Conflicts of interestThe authors declare no conflicts of interest.
We are very grateful to Suzhou BioNovoGene (http://www.bionovogene.com) for technical supports in the comprehensive analysis of the MS/MS data. This study was supported by the National Natural Science Foundation of China (31360564) and Graduate Research & Innovation Project in Xinjiang Autonomous Region of China (XJGRI2014058).