La atresia pulmonar con comunicación interventricular es una cardiopatía congénita cianótica, severa y rara, de alta complejidad, que se caracteriza por la ausencia de conexión entre el ventrículo derecho y las arterias pulmonares. Coexiste con una comunicación interventricular. El flujo hacia el territorio pulmonar puede realizarse a través del ductus arterioso o de colaterales sistémico-pulmonares. La dificultad de esta cardiopatía viene determinada por los diferentes niveles de interrupción desde el ventrículo derecho hasta el territorio pulmonar, y por la diferencia anatómica de las fuentes del flujo hacia dicho territorio, lo que determina diferentes tipos de abordaje quirúrgico.
The pulmonary atresia with ventricular septal defect is a high-complex severe and unusual cyanotic congenital heart disease, characterized by the lack of connection between the right ventricle and the pulmonary arteries; it coexists with a ventricular septal defect. The blood supply of the lungs is possible due to the presence of the patent ductus arteriosus or the major aortopulmonary collaterals. The severity of this heart defect is determined by the extreme variation of the interruption level from the right ventricle to the pulmonary vessel territory, and the anatomy of the blood supply to the lungs that will determine the surgical management strategy.
La atresia pulmonar con comunicación interventricular (AP+CIV) se define como la ausencia funcional de continuidad entre el ventrículo derecho (VD) y las arterias pulmonares, asociada a un defecto del tabique interventricular que generalmente suele ser perimembranoso. Para muchos autores constituye la forma más severa de la tetralogía de Fallot. Es, por tanto, una cardiopatía cianógena que presenta las siguientes características:
- -
Disminución del desarrollo del VD e hipertrofia asociada.
- -
Atresia de la válvula pulmonar con un componente variable de hipoplasia e incluso atresia de las arterias pulmonares.
- -
La asociación de una CIV grande mal relacionada por cabalgamiento aórtico.
- -
El flujo sanguíneo, por lo tanto, llega a las arterias pulmonares a través del ductus arterioso en la mayoría de los casos, y en otras ocasiones, a través de colaterales aortopulmonares, que se conocen por su sigla en inglés, de major aortopulmonary collateral arteries (MAPCA, «arterias colaterales aortopulmonares mayores»).
- -
Lo que hace especial a esta cardiopatía es lo variados que pueden ser los diferentes niveles de interrupción de la vía pulmonar desde el VD hasta las arterias pulmonares.
Para comprender la doble irrigación de ambos pulmones en esta dolencia, es decir, la que proviene del VD y sus ramas y la que surge de la aorta descendente a través de las colaterales aortopulmonares, es necesario resumir el desarrollo embrionario de la circulación pulmonar. Los arcos aórticos son vasos comunicantes que en el embrión conectan las aortas ventrales con las dorsales, y están contenidos dentro de los arcos branquiales1. Los sextos arcos aórticos, que constituyen el par más caudal, están cercanos a los esbozos pulmonares y se conectan con la vasculatura pulmonar. Posteriormente ambos arcos se van a dividir en una porción medial que forma la parte proximal de las ramas de la arteria pulmonar, y otra distal que origina los conductos arteriosos. La unión de los 2 sextos arcos aórticos, por lo tanto, dará lugar a la confluencia de las ramas pulmonares derecha e izquierda que se juntan con el tronco de la arteria pulmonar resultante de la tabicación del tronco cono del corazón, continuación cefálica del VD2.
Pero también en los esbozos pulmonares se desarrollan conexiones con pequeños vasos que surgen de la aorta descendente, que se conectan con las arterias pulmonares a nivel del hilio pulmonar. Estos vasos se desarrollan y aumentan su función cuando la circulación pulmonar proveniente del VD disminuye o está ausente. En condiciones normales, esta circulación colateral desaparece.
AnatomíaLa anatomía intracardiaca es similar a la de la tetralogía de Fallot con estenosis pulmonar. Existe una CIV anterior mal alineada y un VD hipertrófico, sobre todo a nivel del infundíbulo que generalmente no tiene salida, aunque en ocasiones esta puede existir, siendo en estos casos muy pequeña y difícil de observar.
La dificultad de esta cardiopatía viene determinada por lo diferente que puede ser la interrupción de las arterias pulmonares desde el VD hasta las arterias intrapulmonares. De hecho, puede existir atresia de dichas arterias en cualquier parte de su recorrido. Cuando la zona atrésica incluye la válvula y el tronco pulmonar, puede haber ramas pulmonares derecha e izquierda confluentes y unidas entre sí. Este punto es uno de los fundamentales para abordar el tratamiento quirúrgico.
En los pacientes que presentan arterias pulmonares confluentes, suele persistir un ductus, a través del cual se irrigan todos los segmentos pulmonares. Las arterias pulmonares son confluentes cuando ambas arterias intrapericárdicas presentan continuidad entre sí, exista o no tronco de la arteria pulmonar.
Cuando las arterias pulmonares no son confluentes, el 80% tienen una distribución pulmonar incompleta y cerca de una tercera parte de ellas irrigan menos de 10 segmentos pulmonares3,4. Los segmentos pulmonares que no están conectados a las arterias pulmonares centrales habitualmente son irrigados por colaterales aortopulmonares. El conducto arterioso y las colaterales aortopulmonares pueden coexistir en el mismo paciente, pero excepcionalmente lo hacen en el mismo lecho pulmonar.
El conducto arterioso es habitualmente unilateral y se asocia a arterias pulmonares en más del 80% de los casos, aunque aparecen estenosis en el 35 al 50% de los pacientes. También las MAPCA pueden presentar estenosis localizadas en los sitios de la salida aórtica o en la anastomosis intrapulmonar2.
En función de su irrigación a través del ductus y del número de colaterales existentes, puede haber diferentes grados de hipodesarrollo y alteraciones histológicas en los segmentos pulmonares, siendo el grado de hipoplasia de las ramas pulmonares relación del flujo a través del conducto arterioso y del número de colaterales aortopulmonares.
ClasificaciónLa complejidad de la anatomía de la AP+CIV ha dificultado en gran manera su clasificación. Por ello, varios autores han dividido esta afección.
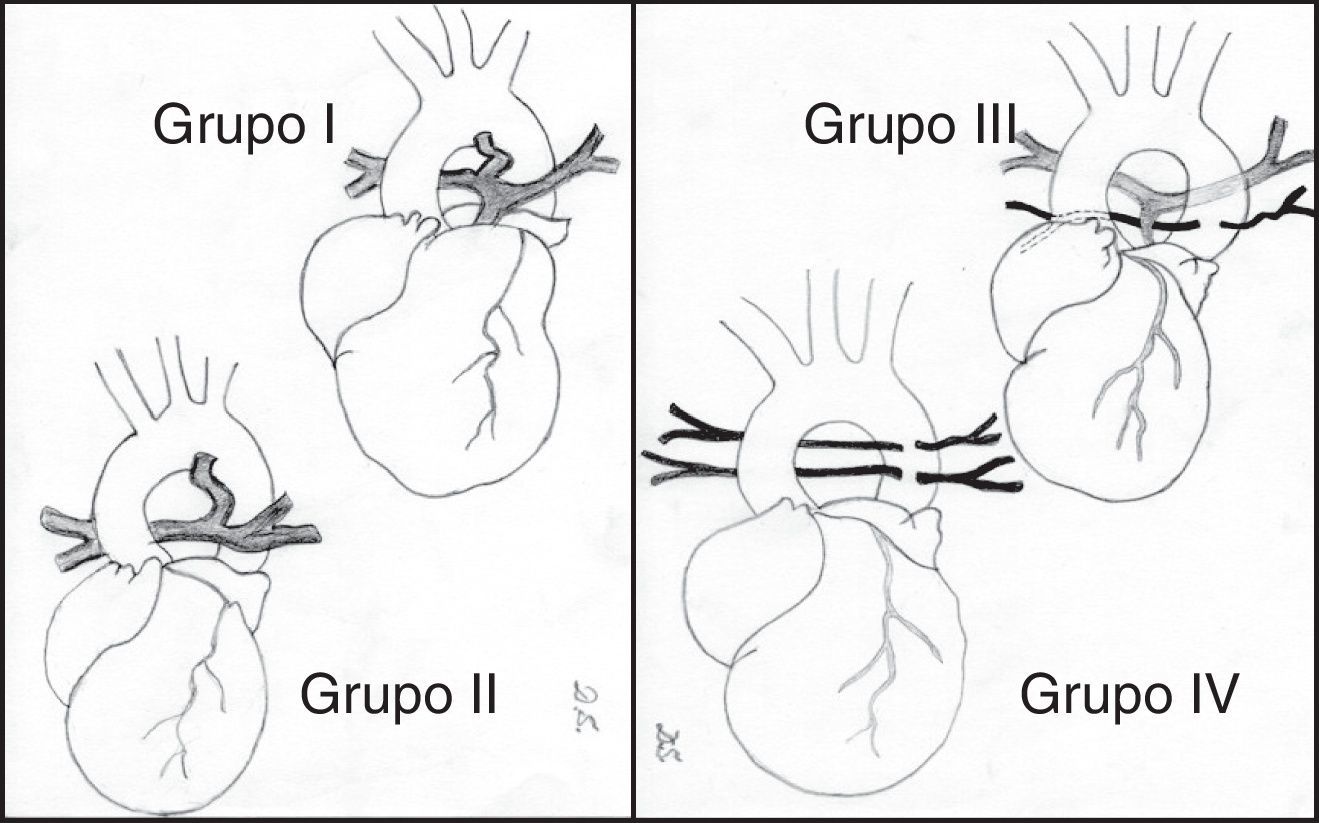
Para Jonas5 (fig. 1), en función de la anatomía de las arterias pulmonares y de la presencia de MAPCA, esta enfermedad se ha dividido en 4 grupos:
- -
Grupo I. La atresia es solo valvular o infundibular.
- -
Grupo II. Existe ausencia del tronco pulmonar, aunque las arterias pulmonares derecha e izquierda son confluentes. La circulación pulmonar es ductus-dependiente.
- -
Grupo III. Las arterias pulmonares son confluentes, aunque severamente hipoplásicas, existiendo además colaterales aortopulmonares.
- -
Grupo IV. Las arterias pulmonares están ausentes y todo el flujo sanguíneo pulmonar5.

Atresia pulmonar con comunicación interventricular. Clasificación. Grupo I: existe una atresia valvular o infundibular. Grupo II: ausencia del tronco pulmonar, aunque las ramas pulmonares derecha e izquierda están en continuidad y la circulación pulmonar depende del ductus. Grupo III: las arterias pulmonares principales están severamente hipoplásicas y la circulación pulmonar es a través de numerosas arterias colaterales aortopulmonares. Grupo IV: las arterias pulmonares centrales están ausentes y todo el flujo pulmonar se recibe a través de colaterales aortopulmonares.
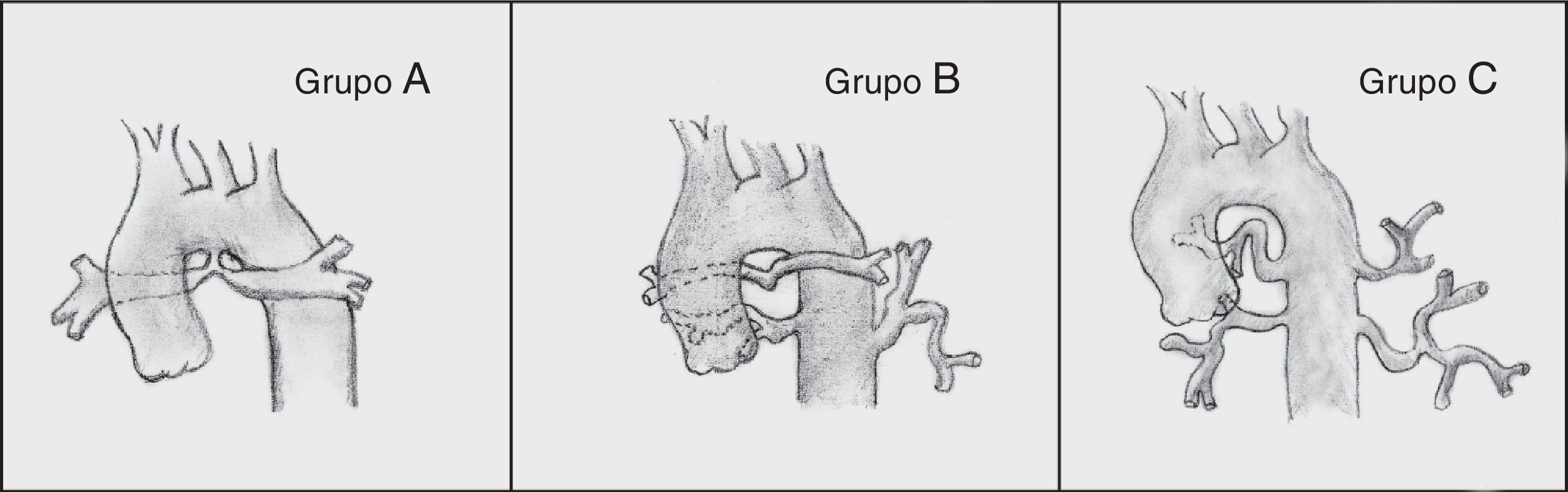
Barbero-Marcial6 describe 3 tipos principales (fig. 2):
- -
Grupo A: incluye los casos en los que todos los segmentos broncopulmonares están irrigados a través de arterias pulmonares centrales. Se divide en 2 subgrupos:
- a)
Cuando los vasos son confluentes y tienen calibre normal o ligeramente disminuido por cierto grado de hipoplasia.
- b)
Cuando las arterias pulmonares centrales tienen estenosis o no son confluentes.
- -
Grupo B: incluye los casos en los cuales algunos segmentos broncopulmonares están irrigados por ramas de las arterias pulmonares y otros reciben flujo a través de colaterales aortopulmonares.
- -
Grupo C: todos los segmentos broncopulmonares reciben flujo a través de colaterales aortopulmonares, ya que no existen arterias pulmonares centrales.

Espectro clínico de la atresia pulmonar con comunicación interventricular según Barbero-Marcial6. Grupo A: todos los segmentos pulmonares están irrigados a través de arterias pulmonares centrales. Grupo B: en estos casos unos segmentos broncopulmonares están irrigados por ramas de la arteria pulmonar y otros reciben flujo a través de colaterales aortopulmonares. Grupo C: todos los segmentos broncopulmonares reciben flujo a través de colaterales aortopulmonares, ya que no existen arterias pulmonares centrales.
Los niños portadores de esta cardiopatía suelen presentar manifestaciones clínicas desde el nacimiento. El signo más evidente es la cianosis. Además, si no existen colaterales aortopulmonares, el cierre del ductus va a producir una hipoxemia severa.
Los lactantes, que debido a la persistencia del ductus o a la presencia de colaterales no presentan clínica florida al nacimiento, progresivamente presentan cianosis y fatiga progresiva, alteración del crecimiento y descompensación cardiaca; todas estas manifestaciones, en relación directa con el mayor o menor flujo pulmonar y el crecimiento y actividad del niño.
DiagnósticoEl diagnóstico de esta enfermedad se realiza mediante ecocardiografía en el período neonatal. Esta prueba diagnóstica es útil sobre todo para definir la anatomía intracardiaca, la presencia de otras lesiones, el tamaño de la arteria pulmonar proximal, la confluencia de las arterias pulmonares intrapericárdicas, así como la posible existencia de anomalías coronarias. También es importante saber si el infundíbulo del VD está o no completamente obstruido, y el tamaño de las arterias pulmonares justo después del origen desde el tronco pulmonar, si existe. Sin embargo, dada la complejidad anatómica de esta enfermedad es necesaria otra prueba diagnóstica que defina perfectamente las colaterales aortopulmonares, la presencia de estenosis en su recorrido, etc. El cateterismo cardiaco y la aortografía deben realizarse también en el período neonatal, para definir perfectamente todas las fuentes de flujo pulmonar, así como para identificar el origen y la significación de todas las MAPCA. También es fundamental la recogida de datos hemodinámicos para valorar la técnica quirúrgica a realizar.
La resonancia magnética, asimismo, resulta de ayuda para el diagnóstico de la AP+CIV, pudiendo mostrar la interrelación entre las colaterales y las arterias pulmonares intrapericárdicas, así como definir la irrigación de los diferentes segmentos pulmonares para planear la cirugía7.
Los nuevos avances en las imágenes de TAC con reconstrucción tridimensional han demostrado su importancia para determinar claramente la anatomía de las colaterales, y así planear la mejor estrategia quirúrgica8.
TratamientoAunque el tratamiento de esta cardiopatía es fundamentalmente quirúrgico, la utilización de prostaglandinas (PGE1) proporcionó un cambio muy importante en la evolución de estos niños9, lo que permite mantener el conducto arterioso permeable hasta el momento de la cirugía.
El objetivo final de la cirugía es cerrar todos los defectos intracardiacos, la desconexión de todas las fuentes de flujo sistémico-pulmonares, conectar el VD a la arteria pulmonar central, y a esta, el mayor número posible de segmentos pulmonares: al menos 14, pero idealmente todos.
La cirugía es obligada en pacientes sintomáticos, bien por disminución del flujo pulmonar, bien por insuficiencia cardiaca. Es necesaria para prevenir la desaparición de colaterales que proveen de flujo a diferentes segmentos pulmonares, así como para prevenir la aparición de hipertensión pulmonar. En los pacientes asintomáticos que no presentan las características previas puede considerarse la cirugía si el riesgo quirúrgico es bajo, la técnica no es difícil y existen altas probabilidades de éxito.
Los procedimientos necesarios para el tratamiento de esta enfermedad dependen del tipo exacto de la anatomía de estos pacientes. Por ejemplo, en aquellos casos que tengan únicamente una atresia de la válvula, puede ser posible abrir esta quirúrgicamente o mediante radiofrecuencia por vía intervencionista, lo que permite restaurar la continuidad entre el VD y el tronco pulmonar10. El éxito de estos procedimientos dependerá del tamaño del VD, del anillo pulmonar, de la válvula tricúspide y de las ramas pulmonares. El cierre de la CIV puede realizarse en un segundo tiempo, o inicialmente si la anatomía es favorable.
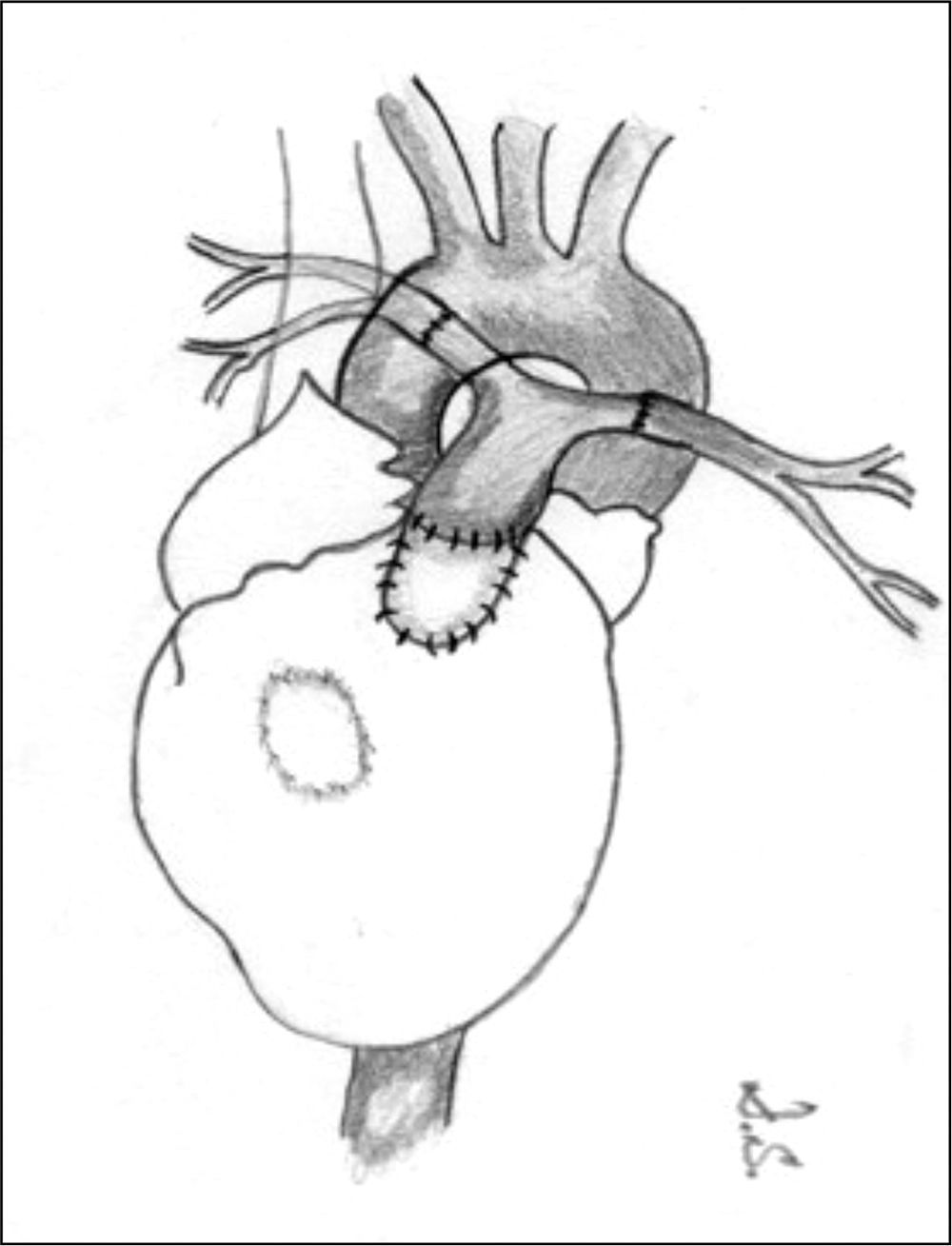
Un porcentaje elevado de pacientes presenta un tracto de salida ventricular derecho muy estrecho, o incluso atrésico, que requiere una reconstrucción, bien sea con la colocación de parches de ampliación, bien con un conducto valvulado o no. Si las arterias pulmonares son confluentes y bien desarrolladas, el conducto puede colocarse desde el tracto de salida hasta las ramas, construyendo un nuevo tronco pulmonar (fig. 3). La CIV puede ser cerrada en la misma cirugía si las arterias pulmonares son de tamaño adecuado.

Las dificultades comienzan cuando las arterias pulmonares son demasiado pequeñas o cuando existe un gran número de MAPCA que irrigan un porcentaje elevado de segmentos pulmonares. En el primer caso, hay que intentar que las arterias pulmonares se desarrollen antes de cerrar la CIV. En ese caso se deben utilizar técnicas paliativas:
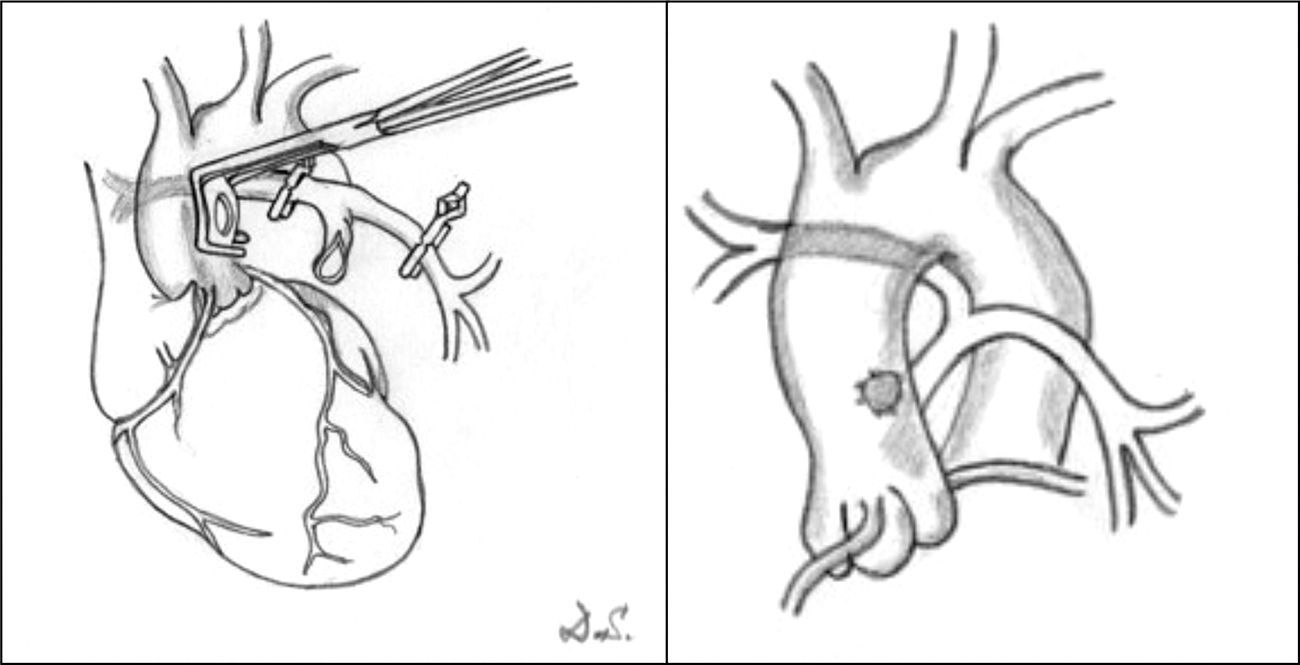
Fístulas sistémico-pulmonares. Se realizan en aquellos pacientes que inicialmente no son candidatos a una reparación y que necesitan mejorar su sintomatología, o como etapa previa a la corrección en una etapa posterior. Lo más frecuente es que estos pacientes presenten hipoxemia severa, ya que necesitan flujo urgente en el lecho pulmonar. Para ello se han utilizado diferentes técnicas de baipás, que conecta la aorta o alguna de sus ramas y el árbol pulmonar. La derivación más utilizada ha sido la de Blalock-Taussig modificada11,12, que se recomienda utilizar en el lado derecho a través de toracotomía o esternotomía media. También puede realizarse una ventana aortopulmonar13 o un baipás central entre la aorta y el tronco pulmonar hipoplásico1, sobre todo en aquellos niños que presentan ramas pulmonares pequeñas (fig. 4).

Hemicorrección. Consiste en la conexión entre VD y arteria pulmonar con parche, conducto sintético u homoinjerto manteniendo abierta la CIV. Proporciona mayor crecimiento de las ramas pulmonares y permite la realización de técnicas percutáneas sobre estas14.
En la mayoría de los pacientes estas técnicas proporcionan un crecimiento suficiente de las arterias pulmonares como para llevar a cabo el cierre de la CIV en una segunda etapa. En otras ocasiones aparecen estenosis localizadas en las ramas, que pueden ser dilatadas mediante procedimientos percutáneos.
La complejidad quirúrgica aumenta cuando existen colaterales aortopulmonares. Cuando estas van directamente a las ramas pulmonares, su oclusión quirúrgica o percutánea es suficiente para evitar el aumento progresivo de la presión pulmonar. Si el paciente va a ser intervenido es conveniente su oclusión mediante cateterismo previo a la cirugía, comprobando que no se produce hipoxemia severa15. Sin embargo, cuando algún segmento pulmonar está irrigado solo por MAPCA, su oclusión no puede llevarse a cabo, ya que produciría un infarto pulmonar a ese nivel. En este caso, durante la cirugía deben desconectarse de la aorta y unirse al sistema arterial pulmonar. Estas técnicas quirúrgicas se conocen como técnicas de unifocalización.
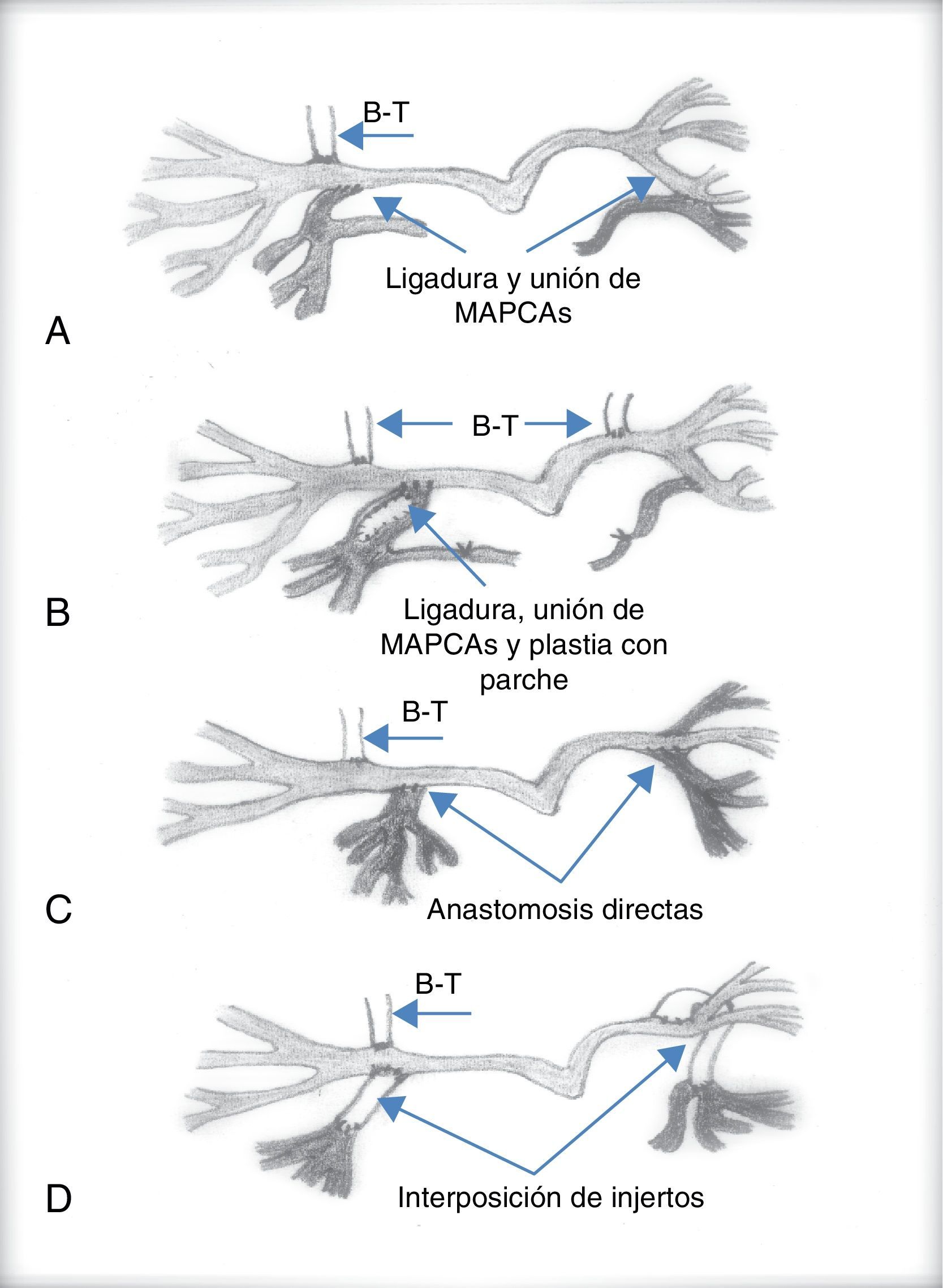
Las técnicas de unifocalización pretenden conectar al menos 14 o 15 segmentos de los 20 que forman el árbol pulmonar entre sí, sin estenosis que impida el flujo a su través16–18. El diagnóstico angiográfico es imprescindible para definir el tamaño y la distribución de las MAPCA. Con este mapa angiográfico el cirujano debe establecer las anastomosis necesarias entre ellas y/o las arterias pulmonares centrales cuando existan, para conectarlas entre sí, y desconectarlas de la aorta de manera que se forme una fuente única que proporcione sangre a un número suficiente de segmentos pulmonares. El procedimiento quirúrgico puede ser complejo, por lo que la principal decisión a la que se enfrenta el cirujano radica en la realización de este procedimiento en una o varias etapas (figs. 5 y 6).
- -
Inicialmente, la cirugía correctora de la mayoría de los pacientes se llevaba a cabo en varias intervenciones. En un principio se realizaba una fístula sistémico-pulmonar, que además de proporcionar flujo al territorio pulmonar, permitía el crecimiento de las ramas pulmonares y del niño, y luego se iban realizando anastomosis de las colaterales aortopulmonares en varias etapas mediante toracotomías sucesivas que permitían unifocalizar la circulación pulmonar y, por último, mediante esternotomía media se cerraba el defecto interventricular y se reconstruía el tracto de salida ventricular derecho y su conexión con las arterias centrales pulmonares. Evidentemente cada uno de estos procedimientos lleva implícita una morbimortalidad no desdeñable. Por otra parte, cuanto más tiempo permanezcan las colaterales irrigando los segmentos pulmonares, más posibilidades hay de que estas sufran estenosis progresivas, y se desarrolle un daño irreversible en el lecho vascular pulmonar.
- -
Más recientemente se tiende a realizar la corrección en una sola etapa17, intentando que el lecho vascular pulmonar pueda aceptar todo el gasto cardiaco sin una presión en el VD superior al 75% de la sistémica19–21. La conveniencia de cerrar o no el defecto interventricular durante esta cirugía constituye una decisión compleja. Cuando la cirugía se realiza en diferentes etapas se han manejado diferentes índices prequirúrgicos para contestar a esta pregunta, entre los que destacan los siguientes:
- a)
Índice de Nakata22: fórmula que permite objetivar el tamaño de las arterias pulmonares en enfermedades congénitas. Este índice relaciona el área de ambas arterias pulmonares centrales justo antes de su bifurcación (área de sección transversal APD+API en mm2/superficie corporal en m2). Un índice superior a 200mm/m2 permitiría el cierre de la CIV.
- b)
Total new pulmonary artery index23: áreas de sección transversal de las APD+API+MAPCA unifocalizables en mm2/superficie corporal en m2. Valores entre 150 y 200mm2/m2 se consideran críticos. Por encima de 200 se considera un valor seguro para cerrar la CIV.
- c)
Índice de McGoon: se calcula dividiendo la suma de los diámetros de la APD y la API entre el diámetro de la aorta a nivel del diafragma. El valor medio en los sujetos normales es de 2,1. Valores superiores a 1,2 se asocian con un posoperatorio aceptable. Por debajo de 0,8 se contraindica el cierre de la CIV.
.")
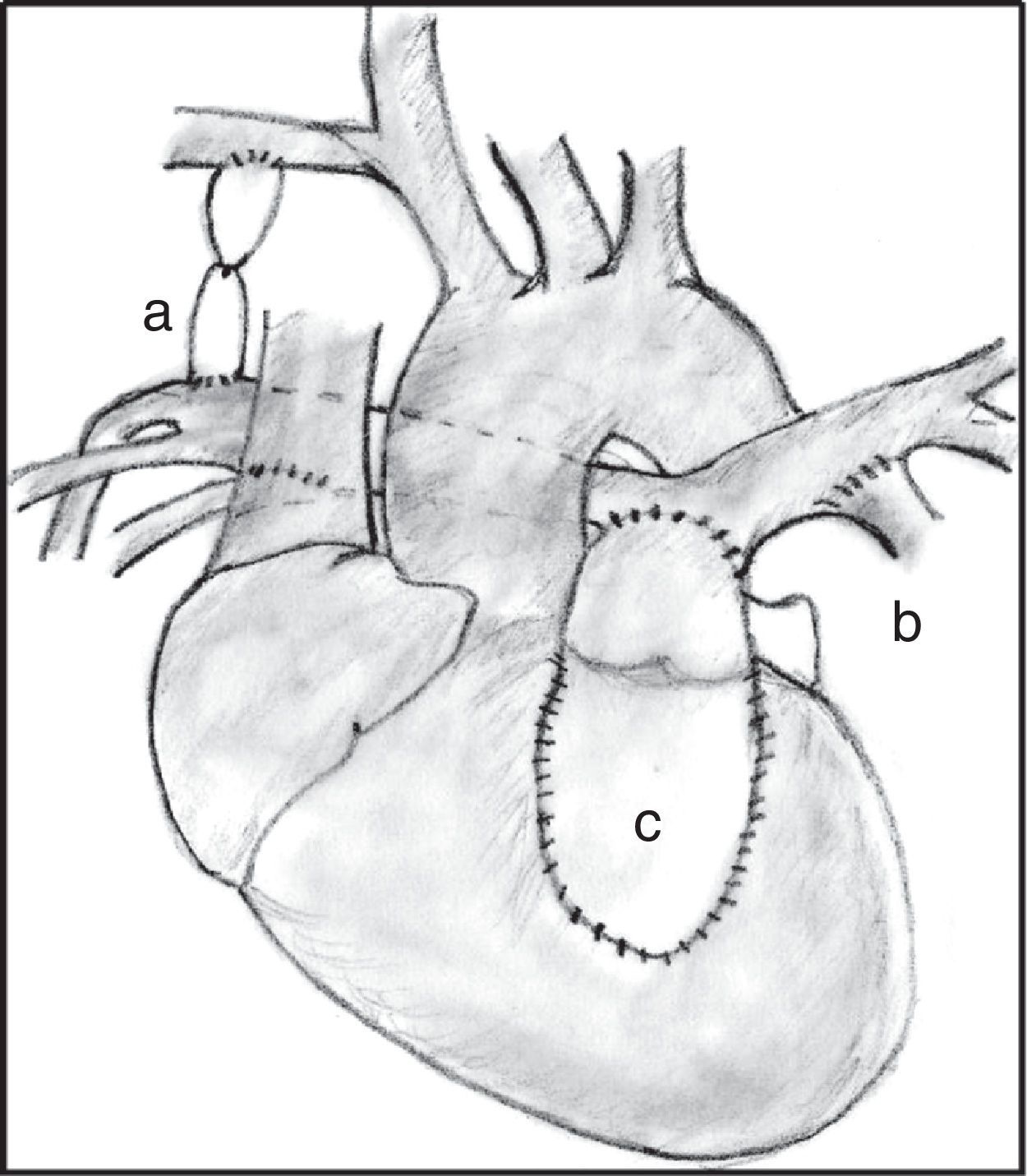
, las conexiones entre colaterales y las ramas pulmonares (b), y el conducto entre el ventrículo derecho y las ramas unifocalizadas (c).")
Cirugía correctora en paciente con ramas pulmonares confluentes y arterias colaterales aortopulmonares mayores. Se puede observar la fístula sistémica-pulmonar cerrada (a), las conexiones entre colaterales y las ramas pulmonares (b), y el conducto entre el ventrículo derecho y las ramas unifocalizadas (c).
La decisión de cerrar la CIV es muy importante, ya que un cierre inapropiado deriva en hipertensión en cavidades derechas por un gradiente alto entre el VD y las arterias pulmonares, y un fallo precoz del mismo. Y por otra parte, dejar la CIV abierta puede dar lugar a un hiperaflujo pulmonar e insuficiencia cardiaca. Por ello, algunos autores23,24 proponen un método durante la cirugía, bajo circulación extracorpórea, que relaciona el flujo en la arteria pulmonar con la presión en la aurícula izquierda. Cuando se alcanza un flujo en las arterias pulmonares unifocalizadas de hasta 3,75l/min/m2, y la presión media pulmonar medida en la aurícula izquierda no supera los 25mmHg, se considera que podría cerrarse la CIV. En caso de duda, puede realizarse una fenestración de 3 a 5mm en el parche de cierre del defecto. Este orificio permite la descarga del VD en caso de que las resistencias pulmonares sean demasiado altas. Posteriormente, si estas disminuyen, la fenestración puede cerrarse por vía percutánea o quirúrgica.
ResultadosLa cirugía en los pacientes con un sistema arterial pulmonar adecuado y sin MAPCA tiene unos resultados similares a los de la tetralogía de Fallot. Sin embargo, la mortalidad precoz en aquellos pacientes que requieren procedimientos complejos de unifocalización puede alcanzar, según los estudios, hasta un 35%25.
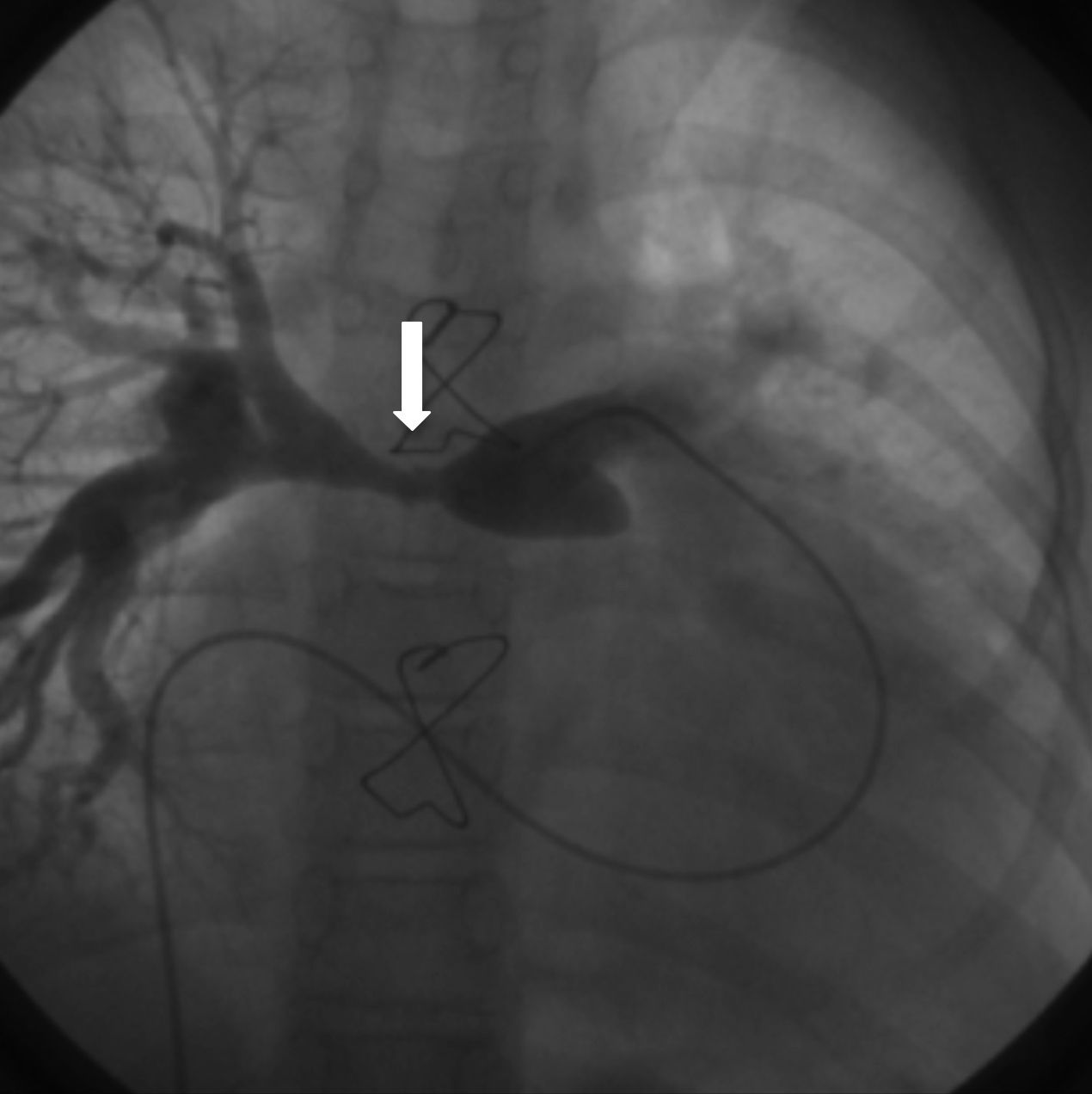
Respecto a la supervivencia a largo plazo, son varios los parámetros que influyen en ella. Resulta complicado comparar las distintas series, debido a las diferencias anatómicas entre ellas y a la mezcla de técnicas quirúrgicas que se comparan. La mortalidad tardía generalmente se debe a diferentes causas, pero fundamentalmente a las reintervenciones necesarias, ya sea para cambiar el conducto o la válvula pulmonar26,27, aunque también las arritmias, las estenosis de las ramas pulmonares (fig. 7), el fallo cardiaco, la presencia de complicaciones neurológicas y la endocarditis pueden influir en la supervivencia a largo plazo.

También la edad en el momento de la corrección influye directamente en la incidencia de reoperaciones en estos pacientes. Para aquellos intervenidos con menos de 5 años, solo el 10% permanecen libres de reintervención a los 10 años28.
La historia natural de los pacientes con esta cardiopatía resulta difícil de determinar debido a la gran variedad anatómica que estos pacientes presentan.
En principio, se diferencian 3 tipos de pacientes29:
1. Alrededor del 50% de ellos tiene ramas pulmonares confluentes normales o levemente hipoplásicas; la circulación se realiza a través del ductus, que tiende a cerrarse, por lo que estos pacientes están enseguida muy hipóxicos. Sin tratamiento, el 50% suele morir en los primeros 6 meses de vida, y solo el 10% sobreviven más de un año.
2. El segundo grupo lo constituye aproximadamente el 25% de los pacientes que tiene alguna colateral aortopulmonar que proporciona flujo a los pulmones y evita la cianosis extrema. De ellos, la mitad fallecen antes de los 3 años de vida, y el 90%, antes de los 10 años.
3. Por último, el tercer grupo presenta gran número de colaterales, subcianosis y aumento del flujo pulmonar. Estos pacientes pueden sobrevivir hasta la edad adulta, desarrollando en ocasiones una hipertensión pulmonar severa que deriva en síndrome de Eisenmenger.
ConclusionesLa AP+CIV se caracteriza por la interrupción de la salida de sangre desde el VD hasta el territorio pulmonar, junto con una CIV que generalmente suele ser perimembranosa. La complejidad anatómica de esta dolencia necesita el conocimiento exacto de la enfermedad individual de cada paciente. El manejo quirúrgico es complejo y debe ser individualizado teniendo en cuenta la morfología de la CIV, la procedencia del flujo pulmonar en cada uno de los segmentos pulmonares, y la morfología y el tamaño de las arterias pulmonares y de las colaterales aortopulmonares cuando existen. Los resultados de estudios recientes que intentan la corrección en una sola etapa son alentadores y es razonable pensar que tales resultados irán mejorando durante los próximos años gracias a los avances en las técnicas diagnósticas, quirúrgicas, perioperatorias e híbridas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.