La atresia pulmonar es una cardiopatía congénita compleja caracterizada por la falta de continuidad entre el ventrículo derecho y la arteria pulmonar. Cuando se asocia a comunicación interventricular y arterias colaterales aortopulmonares mayores supone un reto quirúrgico que puede precisar diferentes vías de abordaje en distintos tiempos quirúrgicos, como el caso presentado.
Pulmonary atresia is a complex congenital heart disease characterized by the lack of continuity between the right ventricle and pulmonary artery. Pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries remain as a surgical challenge and may require staged surgical approaches, as in the case presented.
La atresia pulmonar es una cardiopatía congénita caracterizada por la ausencia de continuidad entre el ventrículo derecho (VD) y las arterias pulmonares (AP) y una comunicación interventricular (CIV) no restrictiva. Como consecuencia de esta falta de paso pueden establecerse vías alternativas para abastecer el territorio pulmonar. Estos cortocircuitos se realizan a través del ductus arterioso o en ocasiones, de forma más infrecuente a través de arterias colaterales aortopulmonares mayores (MAPCA) con origen en aorta torácica descendente o arco aórtico, logrando así la llegada de sangre al pulmón1,2.
En términos generales, se trata de una cardiopatía rara y compleja debido a su gran variabilidad anatómica. El principal signo clínico es la cianosis, pero su grado de manifestación depende de la cantidad de sangre que llega al pulmón a través del ductus arterioso y de las arterias colaterales. De esta forma, en algunos pacientes se presenta con cianosis marcada al nacimiento, mientras que otros lo hacen de forma tardía, junto con estancamiento en el crecimiento e insuficiencia cardiaca. Aunque el tratamiento es fundamentalmente quirúrgico, en algunos casos es necesario administrar prostaglandinas que mantengan inicialmente el ductus abierto3.
De forma específica, la atresia pulmonar asociada a una CIV y a MAPCA es extremadamente infrecuente (10/100.000 nacidos vivos), siendo variable su abordaje quirúrgico. A continuación presentamos un caso con estas características que precisó cirugía en 2 tiempos1.
Caso clínicoPaciente nacida a término con ecocardiografía transtorácica al nacimiento que confirma la existencia de atresia pulmonar y una CIV perimembranosa de 7mm, iniciándose tratamiento con prostaglandina E1. Al cuarto día, el cateterismo demostró arco aórtico derecho, la existencia de AP confluentes (2-3mm de diámetro) con presencia de 2 MAPCA, una de ellas originada en la aorta torácica descendente dirigida hacia el lóbulo pulmonar inferior izquierdo y otra del istmo aórtico perfundiendo el lóbulo pulmonar superior derecho. El ductus no se encontraba permeable por lo que se suspendieron las prostaglandinas.
Tras confirmación de hallazgos similares por nuevo cateterismo a los 6 meses de edad, la paciente fue intervenida a los 9 meses de vida mediante esternotomía media. Se objetivó AP derecha de 4,5mm e izquierda de 6,5mm. Se realizó cierre de la CIV con parche de pericardio autógeno y no se consideró necesaria la unifocalización de MAPCA, por lo que se ligaron. Asimismo se implantó un conducto valvulado biológico procedente de vena yugular bovina (Contegra®) de 12mm de diámetro entre la AP y el VD. El tiempo total de clampaje fue de 51min y el de circulación extracorpórea de 137min.
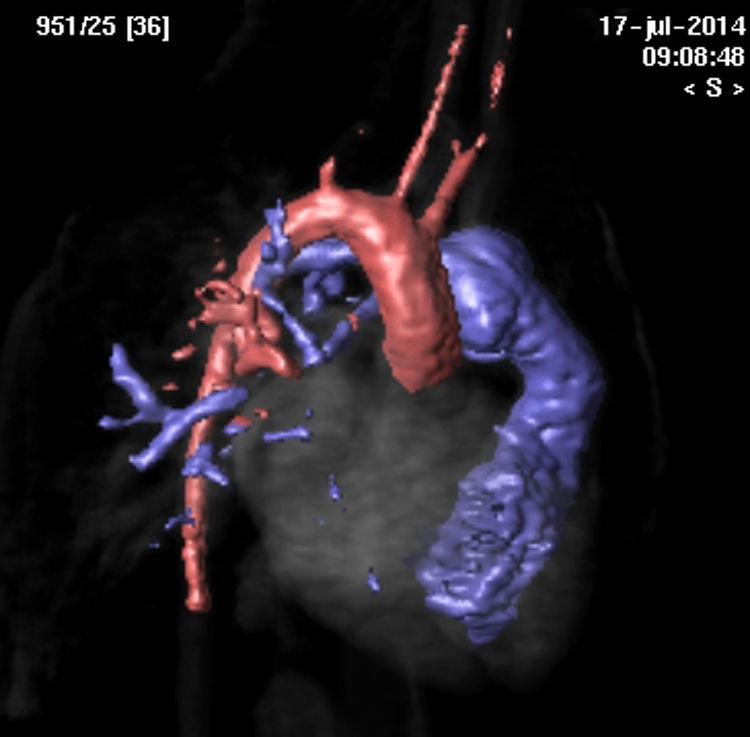
Inicialmente, la paciente permaneció hemodinámica y respiratoriamente estable, aunque en los días posteriores se evidenció una situación de bajo gasto cardiaco, demostrándose en ecocardiografía disfunción severa del VD y persistencia de una MAPCA inferior derecha. Al cuarto día del postoperatorio se realizó cateterismo que confirmó la presencia de 2 MAPCA permeables originadas en aorta descendente, y dirigidas hacia el lóbulo pulmonar inferior derecho, inadvertidas en las pruebas de imagen previas. La realización de una resonancia magnética confirmó los hallazgos anteriormente descritos (figs. 1 y 2). La paciente permaneció estable, y en el plazo de un mes se planeó nuevo cierre quirúrgico de las MAPCA dado el excesivo calibre para su adecuado cierre percutáneo.


Se utilizó como vía de acceso una toracotomía posterolateral derecha a nivel del cuarto espacio intercostal. Tras la disección de la aorta torácica y del arco aórtico derecho se localizaron las 2 MAPCA derechas (fig. 3) y fueron ligadas de forma exitosa. La evolución postoperatoria fue satisfactoria siendo dada de alta a los 10 días sin complicaciones. En el seguimiento a 3 meses permanece asintomática y con adecuada ganancia pondoestatural.
Discusión
La complejidad de la atresia pulmonar con CIV se debe a la gran variabilidad anatómica que obliga a individualizar la estrategia quirúrgica. El cierre de la CIV puede realizarse en un segundo tiempo o inicialmente si la anatomía es favorable, junto con la reconstrucción del tracto de salida del VD, bien con un parche de ampliación o con un conducto valvulado. Si las arterias pulmonares son confluentes y bien desarrolladas, este conducto se dirigirá desde el VD hasta estas ramas construyendo un nuevo tronco pulmonar4,5.
La asociación más infrecuente es la atresia pulmonar con CIV y MAPCA, que se considera aún un reto estratégico. La morfología del árbol pulmonar nativo, ya sean AP confluentes intrapericárdicas (tipo I) o intrapulmonares (tipo II) o no confluentes intrapulmonares (tipo III), así como la distribución y número de MAPCA va a determinar tanto su clasificación anatómica como el pronóstico a largo plazo en estos pacientes, tal como fue descrito por el grupo de Birmingham (Reino Unido) desde el 20046,7.
Cuando las MAPCA van directamente a las ramas pulmonares, su oclusión quirúrgica es suficiente para evitar el aumento progresivo de la presión pulmonar. Sin embargo, cuando algún segmento pulmonar esta irrigado solo por estas colaterales, su oclusión no puede llevarse a cabo ya que produciría un infarto pulmonar a ese nivel. En este caso, es necesario desconectarlas de la aorta y unirlas al sistema pulmonar garantizando así el máximo reclutamiento de los segmentos pulmonares, y construyendo una circulación sistémica y pulmonar independientes. Si bien las técnicas quirúrgicas inicialmente se basaban en derivaciones paliativas, hoy en día se pretenden reconstrucciones más fisiológicas como la unifocalización de MAPCA, la reconstrucción intrahiliar e intrapericárdica y la focalización central hacia el VD8,9.
Nuestro caso es un ejemplo de la complejidad diagnóstica propia de esta enfermedad. No es infrecuente que las MAPCA sufran modificaciones anatómicas desde el nacimiento hasta los 6-9 meses en los que se indica la cirugía. Además, la presencia de ramas pulmonares bien desarrolladas y confluentes permitió que mediante el implante de un tubo valvulado, se restaurase la continuidad entre el VD y la AP. De la misma forma, un tamaño óptimo de ambas AP con adecuada arborización distal, junto con la existencia de circulación dual en los lóbulos inferior izquierdo y superior/inferior derechos, no dependiente exclusivamente del flujo de las MAPCA, hizo innecesaria la unifocalización dada la preservación de la irrigación de un número superior a 15 segmentos pulmonares, permitiendo su ligadura sin complicaciones. Por otra parte, el cierre de arterias colaterales mediante toracotomía posterolateral permite una buena alternativa como vía de acceso, evitando las complicaciones propias de la reesternotomía y permitiendo una visión adecuada de estas MAPCA.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.