




Sr. Director:
Los tumores neuroendocrinos del tracto digestivo forman una entidad clinicopatológica heterogénea potencialmente muy agresiva, que en numerosas ocasiones ha sido confundida con tumores carcinoides y con carcinomas indiferenciados. El carcinoma neuroendocrino es un tumor epitelial raro que comprende un espectro de entidades histopatológicas de alta malignidad y habitualmente de evolución tórpida1,2. Hasta hace poco existía una cierta confusión en cuanto a la nomenclatura de las diversas variedades que definen al carcinoma neuroendocrino. En la actualidad se tiende a unificar criterios histológicos, lo que permite su correcta clasificación. El diagnóstico precoz condiciona un tratamiento efectivo y permite predecir el curso clínico3.

A raíz de haber intervenido recientemente en nuestro hospital a una paciente con un carcinoma neuroendocrino de ciego, analizamos el cuadro clínico, histológico e inmunohistoquímico y revisamos la bibliografía existente al respecto.
Se trataba de una mujer de 76 años de edad con antecedentes de cardiopatía isquémica y hernia diafragmática de Morgagni, que acudió a urgencias por presentar dolor abdominal localizado en hemiabdomen derecho de 2 meses de evolución, tipo cólico, acompañado de estreñimiento y exacerbado en la última semana. En la exploración física destacaba únicamente la presencia de una masa dura, irregular, fija, de 6 cm de diámetro, localizada en el vacío y la fosa ilíaca derecha. El hemograma, bioquímica, estudio de coagulación y ECG eran normales. La radiología de tórax demostraba una masa paracardial derecha compatible con hernia de Morgagni, y la del abdomen era inespecífica. Ingresó en el servicio de cirugía para estudio, donde se realizó una ecografía abdominal que informaba de la presencia de una masa abigarrada en fosa ilíaca derecha de origen digestivo, y un enema opaco que ponía de manifiesto un ángulo hepático y parte derecha del colon transverso de localización intratorácica con paso a través de foramen diafragmático anterior y un defecto de repleción que ocupaba prácticamente la luz del ciego compatible con neoplasia. Fue imposible practicar colonoscopia por mala tolerancia de la paciente.
Se intervino de forma programada a través de una laparotomía media, encontrando una gran hernia de Morgagni que contenía colon ascendente, ángulo hepático y mitad de colon transverso, y una gran tumoración neoplásica en ciego que englobaba anejo derecho e infiltraba retroperitoneo, respetando el uréter. Se practicó hemicolectomía derecha ampliada y anexectomía derecha con anastomosis látero-lateral mecánica y cierre del orificio diafragmático. El postoperatorio transcurrió sin alteraciones.
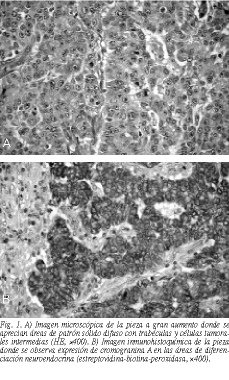
El estudio anatomopatológico macroscópico informaba de tumoración nodular en ciego de 8 * 6 cm, ulcerada centralmente, de crecimiento expansivo con infiltración de capas musculares y grasa pericólica, de la que se aislaron 12 ganglios linfáticos, algunos de ellos de 1 cm de diámetro. Adherido a la misma se identificaron apéndice cecal, anejo uterino, trompa con quiste en tercio proximal y ovario. La descripción microscópica de la pieza tumoral fue un carcinoma neuroendocrino de ciego con áreas de adenocarcinoma pobremente diferenciado, que se extendía a grasa subyacente y respetaba ganglios linfáticos, apéndice, anejo uterino y márgenes quirúrgicos. Se observó un patrón sólido difuso, trabecular o en nidos con células de tamaño intermedio que demostraban núcleos discretamente polimorfos de cromatina fina con micronucleolos y citoplasmas poligonales definidos (fig. 1A). En áreas se apreciaba diferenciación mucosecretora con vacuolas citoplásmicas PAS diastasa positivas. El índice proliferativo celular era elevado, con frecuentes mitosis atípicas, apoptosis y áreas de necrosis. La inmunohistoquímica demostraba positividad de las células tumorales a citoqueratina AE1, CAM 5.2 y cromogranina A en las áreas de diferenciación neuroendocrina (fig. 1B). El tipo era predominantemente neuroendocrino y el subtipo celular correspondía a la variedad de células intermedias.
Sometida a control y revisiones en consultas externas, la enferma se encuentra sin signos de recidiva tumoral a los 20 meses de la intervención quirúrgica, y en la actualidad sigue tratamiento adyuvante combinado con 5-fluorouracilo y ácido folínico.
El tracto gastrointestinal posee el mayor número de células neuroendocrinas del organismo. Éstas son capaces de sintetizar, almacenar y secretar una gran variedad de neuroaminas, péptidos y diversas sustancias que actúan como hormonas o como reguladoras de crecimiento y diferenciación. La proliferación neoplásica de estas células es más frecuente en el apéndice vermiforme, íleon y recto, seguidos del esófago, estómago, duodeno, yeyuno y colon. En el pasado el término carcinoide, modificado algunas veces con palabras como atípico, pleomórfico o maligno, era utilizado para referirse a todo tipo de tumores neuroendocrinos. Estudios inmunohistoquímicos de algunos carcinomas colorrectales indiferenciados o pobremente diferenciados demostraron áreas de diferenciación neuroendocrina, lo que obligó a replantear la definición de esta entidad, discutir su histogénesis y estudiar sus diversas variedades histopatológicas1,3,4. Gould y Chejfec publican en 1978 la primera serie de carcinomas neuroendocrinos confirmada estructural e histológicamente5, y desde entonces han sido numerosas las observaciones puntuales o en serie de esta entidad.
El carcinoma neuroendocrino de colon y recto comprende un grupo heterogéneo de tumores con evidencia de diferenciación neuroendocrina basada en el análisis histológico o inmunohistoquímico, que suponen únicamente del 1 al 4% de todas las neoplasias malignas de esta localización. Si bien en un principio se consideró que tenían un relativo buen pronóstico, estudios posteriores corroboraron el carácter más agresivo y la extensión metastásica temprana de estos tumores respecto a los cánceres colorrectales de origen exocrino. En las series revisadas no se ha encontrado una distribución predominante por sexos, la edad media de presentación suele estar entre la sexta y séptima décadas, y la localización más frecuente suele ser ciego, recto y sigma1,3,4,6. Las teorías y estudios más recientes apoyarían el concepto de la célula multipotencial (stem cell) en el interior de la mucosa del tracto gastrointestinal, con capacidad para diferenciarse en diversas direcciones y con posibilidad para originar tumores de diferentes estirpes2,7.
Estos tumores han sido divididos en tres variedades según el patrón histológico identificado: neuroendocrino puro, predominantemente neuroendocrino, y tumor con igual expresión neuroendocrina que exocrina1. También ha sido identificada la variedad de carcinoma con diferenciación multidireccional, en la cual habría áreas de adenocarcinoma, de carcinoma de células escamosas y de diferenciación neuroendocrina, tratándose de una variedad muy infrecuente y altamente agresiva8. Por otra parte, se han descrito tres subgrupos según el tipo celular predominante: tumor de células pequeñas, de células intermedias y de células bien diferenciadas, que se distinguirían por el índice de actividad mitótica, pleomorfismo y grado de necrosis, lo que condicionaría el pronóstico de cada grupo6,3. En nuestro caso clasificaríamos el tumor dentro de la variedad predominantemente neuroendocrina y en el subgrupo de células intermedias.
Los diversos tipos de carcinoma neuroendocrino no poseen diferencias significativas en cuanto a la clínica de presentación con el adenocarcinoma de colon, y suele ser dolor abdominal, alteraciones del hábito intestinal, hematoquecia, hemorragia oculta en heces o masa abdominal, no habiendo normalmente síntomas de síndrome paraneoplásico, carcinoide ni anormalidades metabólicas4,6. La diseminación se realiza vía linfática y hematógena, siendo el hígado un lugar usual y temprano de localización de metástasis. El subtipo de células pequeñas es la variedad que metastatiza con mayor asiduidad, sobre todo en el hígado, pero también lo hace en hueso, pulmón y peritoneo. A veces se ha iniciado incluso como enfermedad metastásica diseminada y ha sido durante la realización de la necropsia donde se ha descubierto el tumor primario6,7. El diagnóstico diferencial se realizará fundamentalmente con el linfoma, carcinoma atípico y carcinoide, pero en ocasiones la diferenciación es imposible con las técnicas histológicas convencionales, siendo necesario recurrir a técnicas inmunohistoquímicas o ultraestructurales2.
El carcinoma neuroendocrino de colon y recto es una neoplasia altamente agresiva, y la mayoría de casos suelen presentar un pronóstico infausto. De hecho, una gran cantidad de tumores han metastatizado en el momento del diagnóstico, en contraste con el adenocarcinoma de colon y recto, donde sólo lo hacen el 25%3. Tienen peor pronóstico la variedad de carcinoma neuroendocrino puro y el subtipo de células pequeñas, presentando éste mucha similitud en su historia natural con el carcinoma de pulmón variedad oat cell, seguidos del de células intermedias6,7. La supervivencia estará condicionada por el estadio tumoral en el momento del diagnóstico, pero tras revisar la mayoría de series publicadas hemos comprobado que se trataba de neoplasias en estadios avanzados, C y D de Dukes; el tiempo medio de supervivencia se sitúa entre 6 y 15 meses, y en aquellos pacientes con enfermedad localizada y estadio tumoral precoz se consiguen supervivencias a los 5 años entre el 30 y el 90%1,3,4,6,9.
El tratamiento quirúrgico no difiere del resto de neoplasias colónicas, pero dada la rápida evolución del tumor debe incluir tratamiento coadyuvante con quimio y/o radioterapia. Los citostáticos ensayados irían desde el clásico 5-fluorouracilo, asociado a diversas sustancias hasta la combinación de etopósido y cisplatino, siendo éste un régimen de mayor toxicidad10. Si se producen manifestaciones de síndrome carcinoide metastásico pueden ser tratadas con la asociación de 5-fluorouracilo + estreptozotocina3. En cualquier caso, los resultados no son muy halagüeños, y la evolución tórpida suele ser la norma.

