





Introducción. Presentamos un estudio prospectivo cuyo objetivo es determinar la frecuencia de cáncer de colon hereditario no asociado a poliposis (CCHNP), en los casos incidentes de cáncer colorrectal, en dos áreas hospitalarias de la provincia de Jaén.
Pacientes y métodos. El objetivo del trabajo fue determinar la frecuencia de CCHNP. El estudio se llevó a cabo en la población de referencia de dos áreas hospitalarias de la provincia de Jaén (234.500 habitantes), y tuvo una duración de 2 años. Para identificar las familias con CCHNP nos hemos basado en la historia familiar de los pacientes diagnosticados de cáncer colorrectal. Para establecer el diagnóstico de CCHNP se han aplicado los criterios definidos por el International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (criterios de Amsterdam).
Resultados. Hemos estudiado la historia familiar de 173 pacientes con cáncer colorrectal. Tres familias cumplían todos los criterios diagnósticos de CCHNP. Otras dos familias presentaban una clara asociación familiar de cáncer colorrectal, aunque no cumplían de forma rigurosa los criterios de Amsterdam (probable CCHNP).
Conclusiones. En la población estudiada, la frecuencia de CCHNP se sitúa entre el 1,7% (CCHNP confirmado) y el 3% (incluyendo los casos probables) de los cánceres colorrectales.
Introduction. The aim of this prospective study was to determine the frequency of hereditary non-polyposis colon cancer (HNPCC) in patients with colorectal cancer in two health regions of the province of Jaen (Spain).
Patients and methods. The study was carried out in the reference population of two health service areas in the province of Jaen (234,500 inhabitants) for 2 years. The family history of patients diagnosed with colorectal cancer was used to identify families with HNPCC. The criteria defined by the International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (Amsterdam criteria) were used to establish a diagnosis of HNPCC.
Results. The family histories of 173 patients with colorectal cancer were studied. Three families met all the diagnostic criteria for HNPCC. Another two families showed a clear familiar association with colorectal cancer but did not rigorously fulfill the Amsterdam criteria (probable HNPCC).
Conclusions. In the study population, the frequency of HNPCC was between 1.7% (confirmed HNPCC) and 3% (including probable cases) of colorectal cancers.
Introducción
El cáncer de colon hereditario no asociado a poliposis (CCHNP) o síndrome de Lynch se caracteriza por la aparición de cáncer colorrectal en pacientes menores de 50 años, localización preferente en el colon derecho, alta incidencia de cáncer metacrónico, asociación con neoplasias extracolónicas y transmisión hereditaria autosómica dominante1.
La identificación de familias con síndrome de Lynch tiene una gran trascendencia tanto clínica como epidemiológica debido, en primer lugar, a la importancia de la detección precoz en los miembros asintomáticos de estas familias, con alto riesgo de padecer cáncer colorrectal y, en segundo lugar, a la alta incidencia de cáncer metacrónico en pacientes tratados de cáncer colorrectal.
Actualmente, el único método eficaz para identificar las familias con CCHNP es la realización de una historia familiar detallada en los pacientes con cáncer colorrectal.
Presentamos un estudio prospectivo cuyo objetivo es la identificación de familias afectadas de CCHNP y determinar la frecuencia de este síndrome en los casos incidentes de cáncer colorrectal en la población correspondiente a dos áreas hospitalarias de la provincia de Jaén.
Pacientes y métodos
En julio de 1996 iniciamos un estudio prospectivo cuyo objetivo era determinar la frecuencia de cáncer de colon hereditario no asociado a poliposis, definida como la proporción de casos de cáncer colorrectal cuya historia familiar cumple los criterios diagnósticos establecidos por el International Collaborative Group on Hereditary Non-polyposis Colorectal Cancer. La población de referencia es la correspondiente a las áreas hospitalarias de los hospitales Princesa de España de Jaén y San Agustín de Linares, con un total de 234.500 habitantes, siendo los sujetos del estudio los pacientes diagnosticados de novo de cáncer colorrectal en estos 2 hospitales. La duración del estudio fue de 2 años.
Proceso de identificación de familias con CCHNP
Para identificar a las familias con CCHNP nos hemos basado en la historia familiar de los pacientes diagnosticados de cáncer colorrectal. Durante los 2 años de duración del estudio, todos los pacientes con cáncer colorrectal fueron entrevistados por un cirujano que investigó los antecedentes familiares referidos a enfermedades tumorales en los familiares de primer grado. En aquellos casos en que algún familiar de primer grado había padecido cáncer colorrectal, la historia familiar se amplió a los familiares de segundo grado.
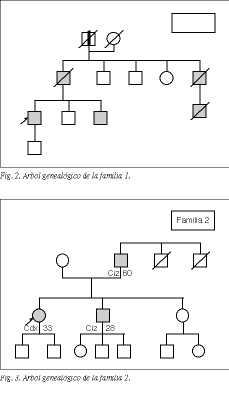
En las familias en las que se detectó una tendencia familiar al padecimiento de cáncer colorrectal, la información aportada por el paciente o los familiares se documentó mediante informes clínicos, que se solicitaron a los hospitales donde fueron tratados los familiares afectados. En algunos casos, cuando no fue posible obtener informes clínicos, la confirmación documental de los diagnósticos se consiguió de la información recogida en el certificado de defunción, previa solicitud al Registro Civil correspondiente. Con los datos obtenidos se han confeccionado los árboles genealógicos, siguiendo la simbología y los códigos que se detallan en la figura 1.
Las familias fueron divididas en 3 grupos, según el número de miembros afectados de cáncer colorrectal en el momento de realizar la historia familiar: a) familias sin antecedentes de cáncer colorrectal; b) familias en las que un familiar de primer grado del "paciente inicio" había padecido cáncer colorrectal, y c) familias con tres o más miembros afectados de cáncer colorrectal.
Criterios diagnósticos de CCHNP
Para establecer el diagnóstico de CCHNP se han aplicado los criterios establecidos por el International Collaborative Group on Hereditary Non-polyposis Colorectal Cancer (criterios de Amsterdam): a) al menos tres familiares deben tener confirmación histológica de cáncer colorrectal. Uno debe ser familiar de primer grado de los otros dos; b) deben estar afectadas dos generaciones sucesivas, y c) uno de los familiares debía tener menos de 50 años cuando fue diagnosticado de cáncer colorrectal.
Resultados
En los 2 años de duración del estudio se diagnosticaron 179 nuevos casos de cáncer colorrectal en la población de referencia, lo que supone una incidencia bruta de 76 por 100.000 habitantes en 2 años. Entre los pacientes, 115 fueron varones y 64 mujeres. La edad media fue de 63 años, con un rango entre los 24 y los 91 años.
En 6 casos no se investigaron los antecedentes familiares de los pacientes, por lo que hemos estudiado las historias familiares de 173 pacientes.

En 146 casos (85%) la historia familiar fue negativa, es decir, no existían antecedentes familiares de cáncer colorrectal. Se hallaron 22 pacientes (12%) con un familiar de primer grado que había padecido cáncer colorrectal, sin que hubiese otros casos entre los familiares de segundo grado. Dentro de este grupo, en 10 familias los miembros afectados pertenecían a 2 generaciones sucesivas.
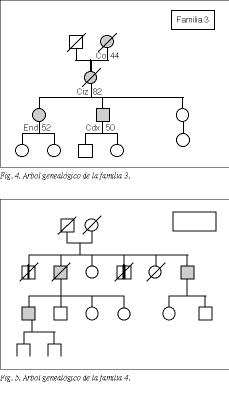
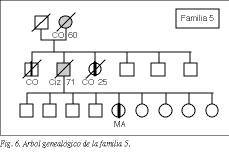
En 5 familias (3%), que hemos designado con los números 1, 2, 3, 4 y 5, se observó una clara tendencia hereditaria a padecer cáncer colorrectal: tres o más miembros habían sido diagnosticados de cáncer colorrectal. Los árboles genealógicos de estas familias se encuentran representados en las figuras 2 a 6.



Dos de estas familias no cumplían de forma estricta los criterios de Amsterdam: una de ellas (familia 4) cumplía todos los criterios excepto que uno de los pacientes fuera menor de 50 años en el momento del diagnóstico (el "paciente inicio" tenía 57 años). En la otra familia (familia 5) no se pudo obtener verificación documental del diagnóstico de uno de los tres familiares afectados de cáncer colorrectal. Por tanto, no satisfacían el requisito de tener la confirmación del diagnóstico en, al menos, tres pacientes. Hemos considerado estos 2 casos como "probable CCHNP".
Tres familias (familias 1, 2 y 3) cumplían todos los criterios diagnósticos de CCHNP. La frecuencia de presentación de este síndrome en la población de referencia es del 1,7% de los cánceres colorrectales.
En dos familias con CCHNP confirmado, algunos de sus miembros presentaron cánceres de localización extracolónica (síndrome tipo II de Lynch), mientras que en la tercera únicamente se diagnosticaron neoplasias de localización colorrectal (síndrome tipo I de Lynch).
Discusión
La primera descripción del síndrome de CCHNP fue realizada por Aldred Warthin en 1895, pero no fue hasta 1963 cuando Lynch et al comunican un exhaustivo estudio de los miembros de una familia con historia de cánceres múltiples, e iniciaron la identificación de familias propensas a desarrollar cáncer co lorrectal en ausencia de poliposis2.
Esta alteración fue conocida inicialmente como "síndrome de cáncer familiar" y, más tarde, en la reunión del International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer, celebrada en Amsterdam en agosto de 1990, se acordó denominarla "cáncer de colon hereditario no asociado a poliposis" o síndrome de Lynch.
En esta misma reunión se definieron los criterios mínimos para establecer el diagnóstico de CCHNP y que se conocen como "criterios de Amsterdam"3.
La principal dificultad para conocer la prevalencia real de este síndrome radica en el hecho de no disponer de biomarcadores o un fenotipo específico para su identificación, y que no puede ser diagnosticado hasta que varios miembros de una familia, en dos generaciones sucesivas, han sido afectados4. Son numerosos los estudios que han intentado identificar las alteraciones genéticas que determinan la aparición del CCHNP. Estas alteraciones, causantes de la susceptibilidad al CCHNP, se han detectado en alrededor del 70% de las familias5. Estudios recientes indican que se producen mutaciones en hMSH2 y hMLH1 en el 50 y el 30% de los casos, respectivamente, siendo mucho menor la frecuencia de mutaciones en hPMS1 y hPMS2 (5%)6. En la búsqueda de marcadores biológicos, otros trabajos han intentado correlacionar la hiperproliferación celular en la mucosa rectal con la predisposición a desarrollar cáncer colorrectal7. Sin embargo, hasta el momento no disponemos de un marcador genético o molecular universal para el diagnóstico de todas las familias con síndrome de Lynch, por lo que el único método eficaz de identificación es la elaboración de una historia familiar detallada y lo más amplia posible8.
La frecuencia de aparición de esta enfermedad es variable y, aunque en algunos estudios es inferior al 0,5%, la mayoría de los autores la sitúan entre el 1 y el 5% de los cánceres colorrectales9-12. Esta disparidad de resultados puede ser debida a diferencias entre las poblaciones estudiadas, pero también está determinada por la desigualdad metodológica entre los distintos estudios. Aunque todos los autores aplican los mismos criterios diagnósticos, no existe uniformidad en los distintos estudios en la selección de pacientes, el protocolo de estudio y el grado de verificación de los diagnósticos2. Para nosotros, obtener la verificación documental de los casos de cáncer colorrectal ha supuesto una dificultad añadida en el proceso diagnóstico, debido a los impedimentos que hemos encontrado, tanto por parte de los hospitales como de los registros civiles a los que hemos demandado información.
En la población que hemos estudiado, la frecuencia de CCHNP se sitúa entre el 1,7%, de casos confirmados y el 3% si incluimos las dos familias que no cumplían de forma estricta los criterios diagnósticos de Amsterdam, de los cánceres co lorrectales.
Creemos que no se debe descartar el diagnóstico de síndrome de Lynch en las dos familias que no cumplen todos los criterios de Amsterdam, y hemos considerado a ambos casos como "probable CCHNP" y son objeto de seguimiento por nuestra parte.
Algunos autores consideran muy restrictivos los criterios de Amsterdam para su aplicación en pequeñas familias9,13. Percesepe et al13 estudian, mediante análisis multivariable, 6 criterios clínicos, todos ellos indicativos de una marcada susceptibilidad genética a padecer cáncer colorrectal: transmisión vertical (uno de los progenitores o un descendiente del "paciente inicio" afectados de cáncer colorrectal u otra localización), agregación familiar (el 50% de los hermanos, o el 50% de los familiares de primer grado, afectados de cáncer de cualquier localización), edad temprana de aparición del cáncer colorrectal, localización en el colon derecho, aparición de múltiples tumores primarios y tipo histológico de adenocarcinoma mucinoso. Estos autores concluyen que cuando se reúnen cuatro de estos criterios en el núcleo familiar la probabilidad de CCHNP es superior al 50%, y que el criterio de mayor valor predictivo en el diagnóstico de CCHNP es la afectación de dos generaciones sucesivas. Cuando se dan estas condiciones no se debe excluir el diagnóstico, aun cuando no se cumplan todos los criterios de Amsterdam. Las 2 familias que hemos considerado como "probable CCHNP" cumplen cuatro de los criterios clínicos antes citados. Además, según estos estudios consideramos que sería aconsejable volver a evaluar, pasados unos años, la historia familiar de aquellos casos en que 2 familiares de 2 generaciones sucesivas hayan sufrido cáncer colorrectal.
De este síndrome se distinguen dos tipos: síndrome de Lynch tipo I, en el que las neoplasias se localizan únicamente en el colon, con las características antes descritas, y síndrome de Lynch tipo II cuando, además, se asocian cánceres de localización extracolónica (endometrio, mama, estómago, vías biliares, etc.). El cáncer de endometrio parece ser el que tiene una asociación más clara con el síndrome de Lynch1. En dos de las familias identificadas se habían diagnosticado cánceres de localización extracolónica, por lo que pertenecen al tipo II de Lynch.
En los pacientes con síndrome de Lynch intervenidos de cáncer colorrectal, la incidencia de cáncer metacrónico es significativamente más elevada que en los casos esporádicos. En los seguimientos a largo plazo realizados en este grupo de pacientes, el riesgo acumulado de padecer una nueva neoplasia (adenoma + carcinoma) colorrectal es del 40% a los 10 años de la intervención por cáncer colorrectal y, en este tiempo, el 25% de los pacientes desarrolló cáncer metacrónico de colon14.
Las implicaciones prácticas de este hecho son evidentes: es obligado realizar un seguimiento estricto de los pacientes intervenidos por padecer CCHNP. Mecklin et al estudiaron durante 7 años a un grupo de pacientes con síndrome de Lynch tratados de cáncer colorrectal y comprobaron que la aparición de cáncer metacrónico en pacientes tratados mediante colectomía segmentaria fue mayor (22%) que en los tratados con colectomía subtotal (11%). En consecuencia, se recomienda la práctica de una colectomía subtotal como técnica quirúrgica de elección para el tratamiento de la neoplasia inicial en pacientes con CCHNP15.
El carácter hereditario autosómico dominante de este síndrome determina que el riesgo teórico de sufrir cáncer colorrectal, en descendientes de familiares afectados, sea del 50%. El alto riesgo de aparición de cáncer colorrectal o de otra localización en miembros asintomáticos de estas familias hace necesario establecer un programa de seguimiento protocolizado de estos familiares16. El seguimiento periódico es una forma de prevención secundaria y conlleva una disminución de la mortalidad por cáncer17.

