




Introducción
La enfermedad de Caroli (EC), descrita inicialmente por Vachel y Stevens en 1906, y posteriormente caracterizada por Caroli en 1958, tiene una base hereditaria incierta y puede asociarse a otras enfermedades de carácter hereditario, en el 60% de los casos, con la enfermedad poliquística renal. Predomina en la mujer, se presenta con afección hepática monolobar en el 15% de los casos y degenera en el 7-24% de los casos en colangiocarcinoma. Su tratamiento ideal en la forma focal es la resección hepática parcial, y está indicado el trasplante hepático cuando se asocia a fibrosis portal o en las formas difusas. Presentamos, por su rareza, 3 casos de EC focal (ECF): 1 caso asociado a EPRAD y otros 2 casos caracterizados por episodios de colangitis de repetición.
Caso clínico 1
Varón de 75 años, diabético tipo 2, hipertenso, con cardiopatía isquémica, trasplantado renal hace 10 años por insuficiencia renal crónica secundaria a EPRAD en actual tratamiento inmunosupresor (ciclosporina 150 mg/día y prednisona 5 mg/día). Diagnosticado de EC hace 14 años, permaneció asintomático hasta la fecha. Ingresa por cuadros de colangitis de repetición. La ecocolangiorresonancia magnética hepática demostró dilatación sacular de la vía biliar intrahepática limitada a lóbulo hepático izquierdo, presencia de colección yuxtahepática y atrofia del lóbulo hepático izquierdo (fig. 1). Con la orientación diagnóstica de colangitis en el contexto de ECF se realizó una lobectomía hepática izquierda sin complicaciones intraoperatorias y se le dio el alta el décimo tercer día postoperatorio. Reingresa 3 meses más tarde, con cuadro séptico y tomografía computarizada (TC) abdominal, que muestra colección intraabdominal indicativa de absceso, por lo que se instaura tratamiento antibiótico de amplio espectro y se realiza drenaje percutáneo de dicho absceso. El paciente se deteriora progresivamente, tiene un fallo multiorgánico y muere.
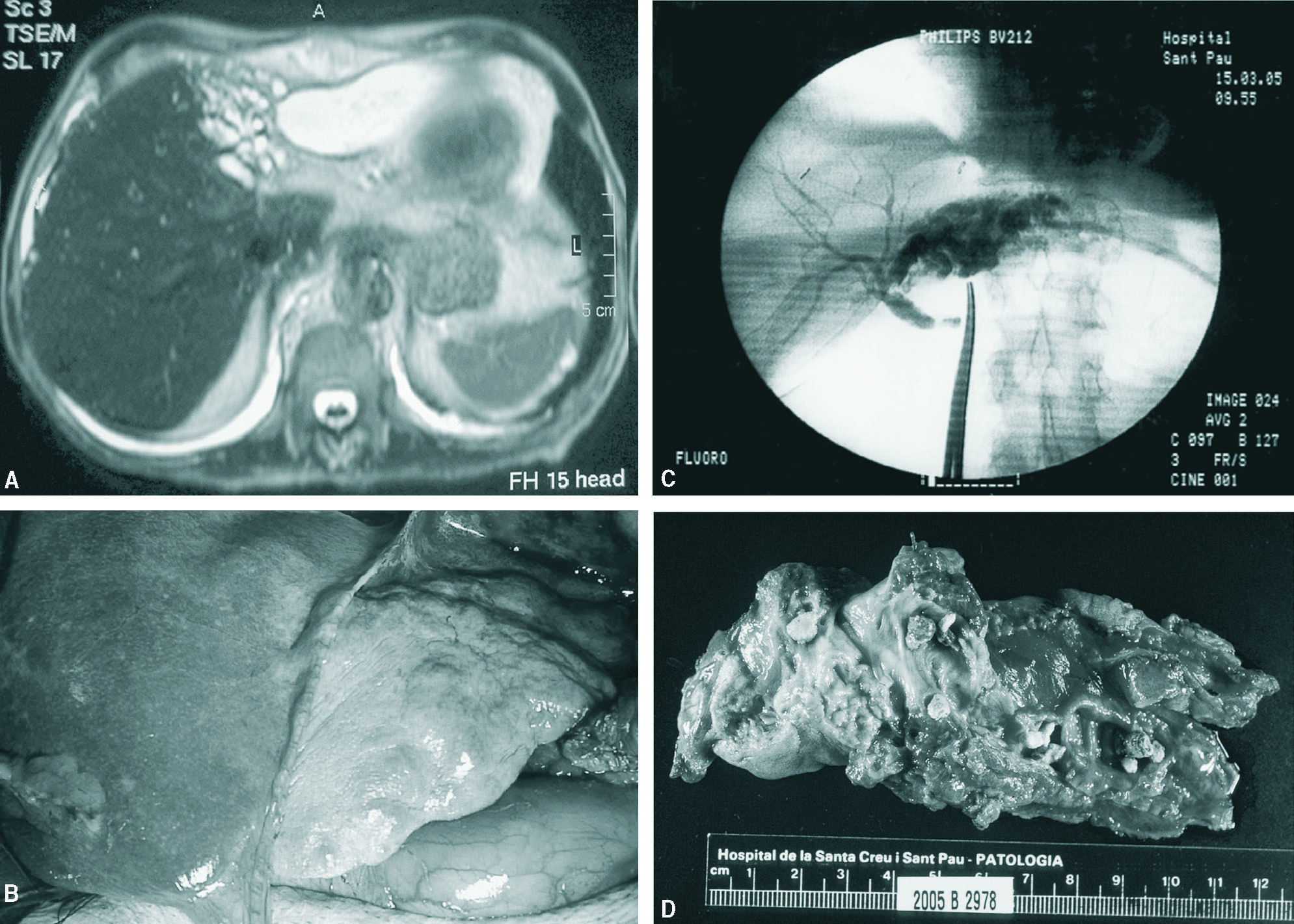
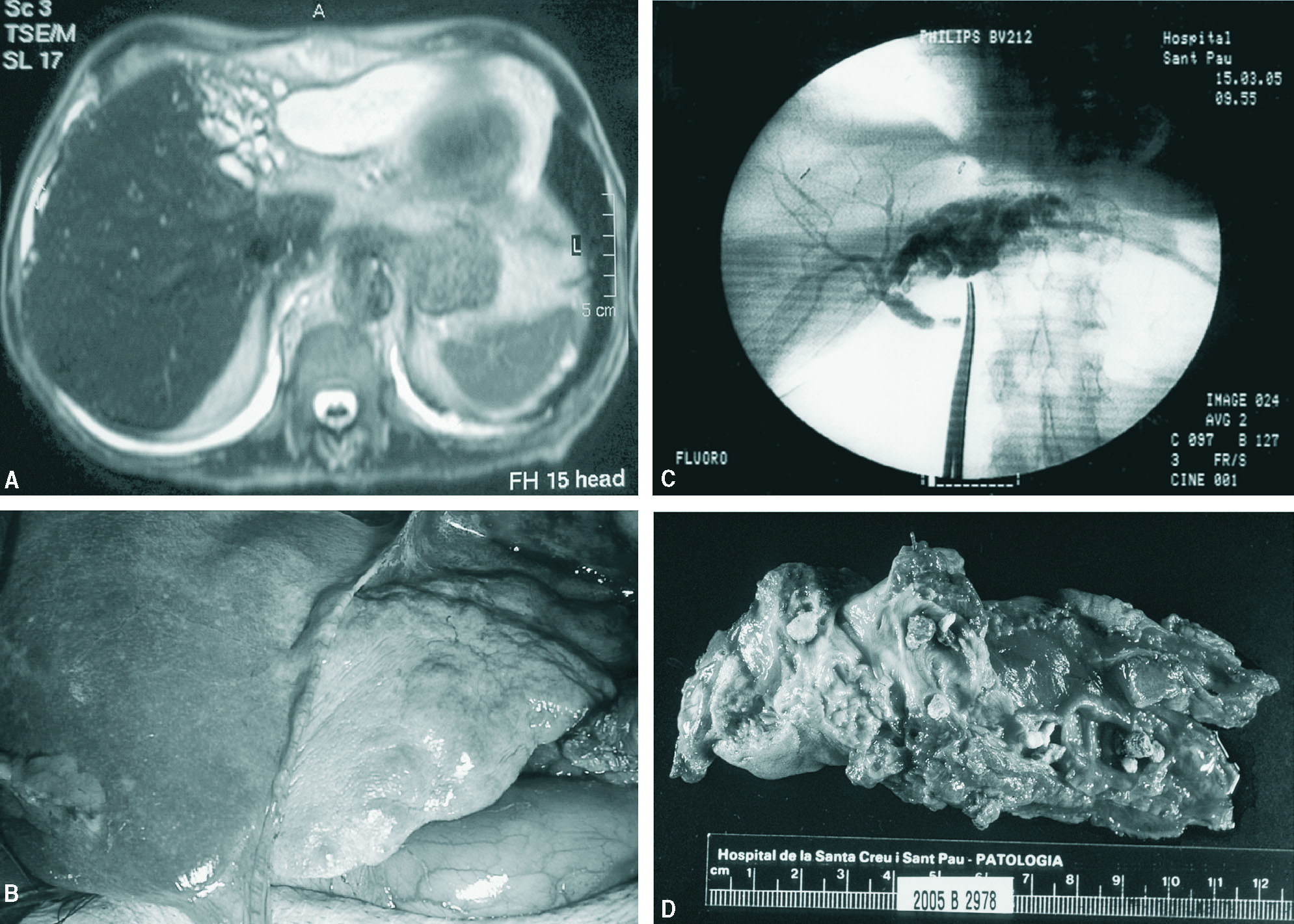
Fig. 1. A: colangiorresonancia magnética. Dilatación sacular de vía biliar izquierda con importante atrofia del lóbulo hepático izquierdo y colección yuxtahepática. B: aspecto macroscópico del lóbulo hepático izquierdo. C: Colangiografía intraoperatoria. D: anatomía patológica macroscópica. Vías biliares intrahepáticas dilatadas con litiasis en su interior.
Caso clínico 2
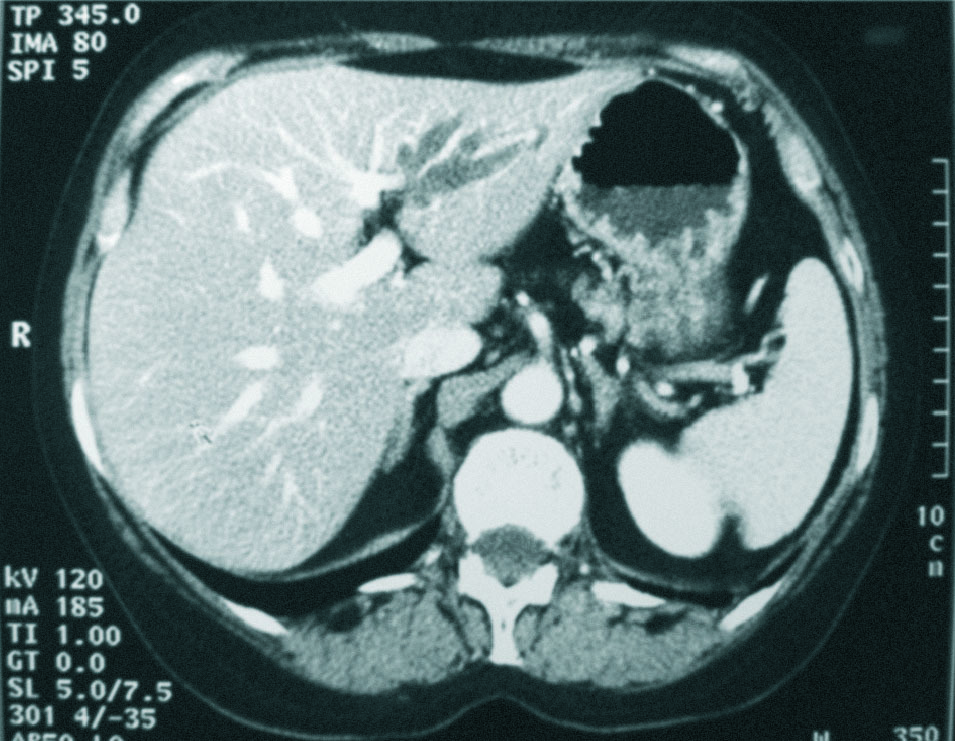
Mujer de 56 años, fumadora, alérgica a sulfamidas, que consulta tras repetidos episodios de cólicos biliares y elevación persistente de GGT. Se realiza colangiorresonancia magnética, que objetiva colelitiasis y una estenosis a nivel del conducto hepático izquierdo irregular, asociada a una dilatación de la vía biliar izquierda de características saculares, con presencia de un defecto de repleción, por una pequeña litiasis. Resto normal (fig. 2). Con la orientación diagnóstica de ECF izquierda, se realiza lobectomía hepática izquierda, con curso postoperatorio sin incidencias. Tras 6 meses de seguimiento la paciente ha permanecido asintomática.
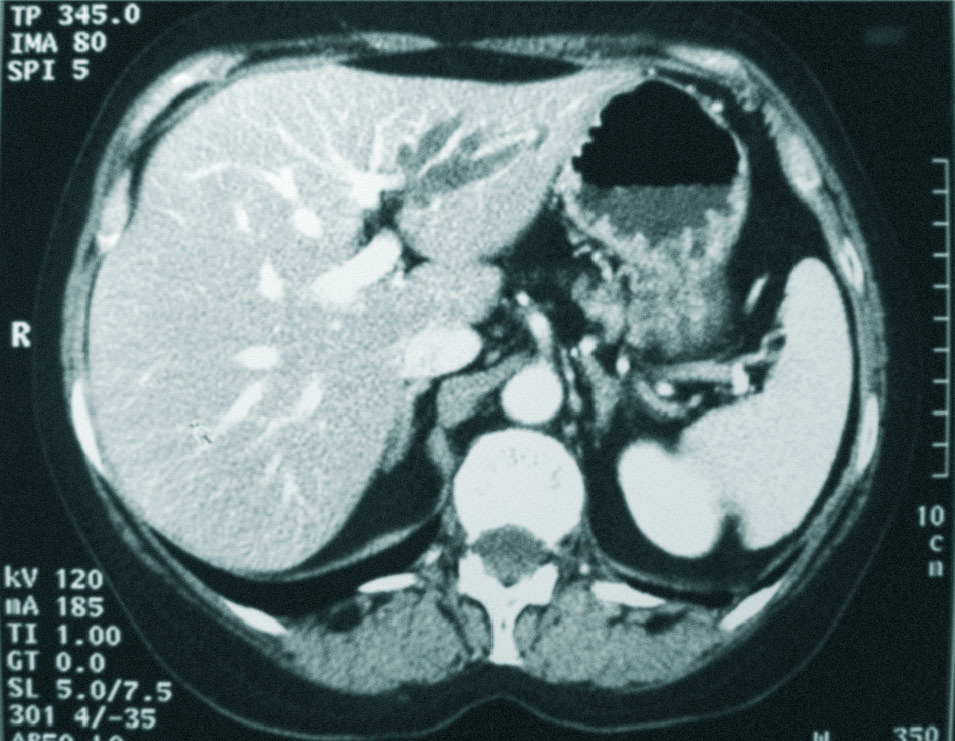
Fig. 2. Tomografía computarizada abdominal con contraste intravenoso fase portal. Dilatación sacular de la vía biliar intrahepática izquierda.
Caso clínico 3
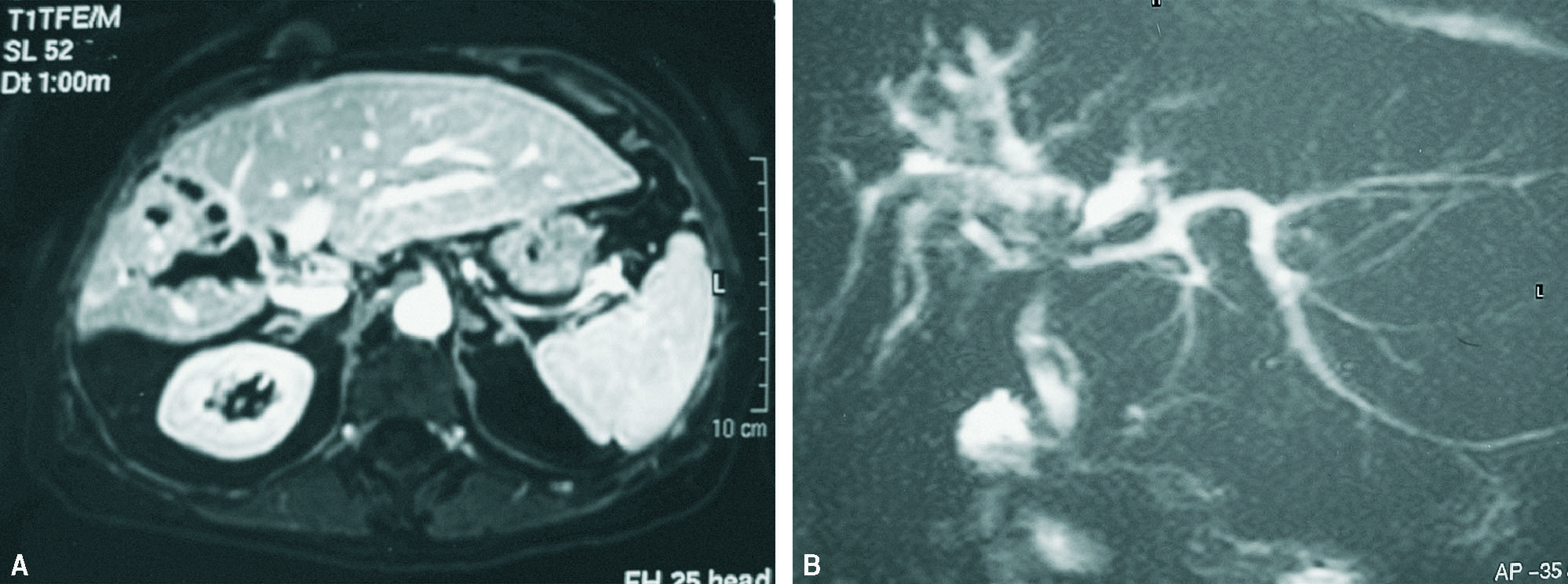
Mujer de 68 años colecistectomizada hace 7 años. Consulta por ictericia obstructiva con colangitis. La ERCP muestra estenosis severa en el origen del conducto hepático derecho, con dilatación sacular supraestenótica y gran número de cálculos retenidos a dicho nivel. Se realiza dilatación quirúrgica de la estenosis y extracción de litiasis con drenaje de Kehr durante 6 meses. Posteriormente, múltiples ingresos sucesivos por colangitis, por lo que finalmente y dado el alto grado de atrofia del LHD, se practica hepatectomía derecha incluyendo la estenosis (fig. 3). Postoperatorio satisfactorio con alta hospitalaria a los 10 días. Controles sucesivos con seguimiento a 1 año, asintomática. El estudio de anatomía patológica, compatible con ECF.
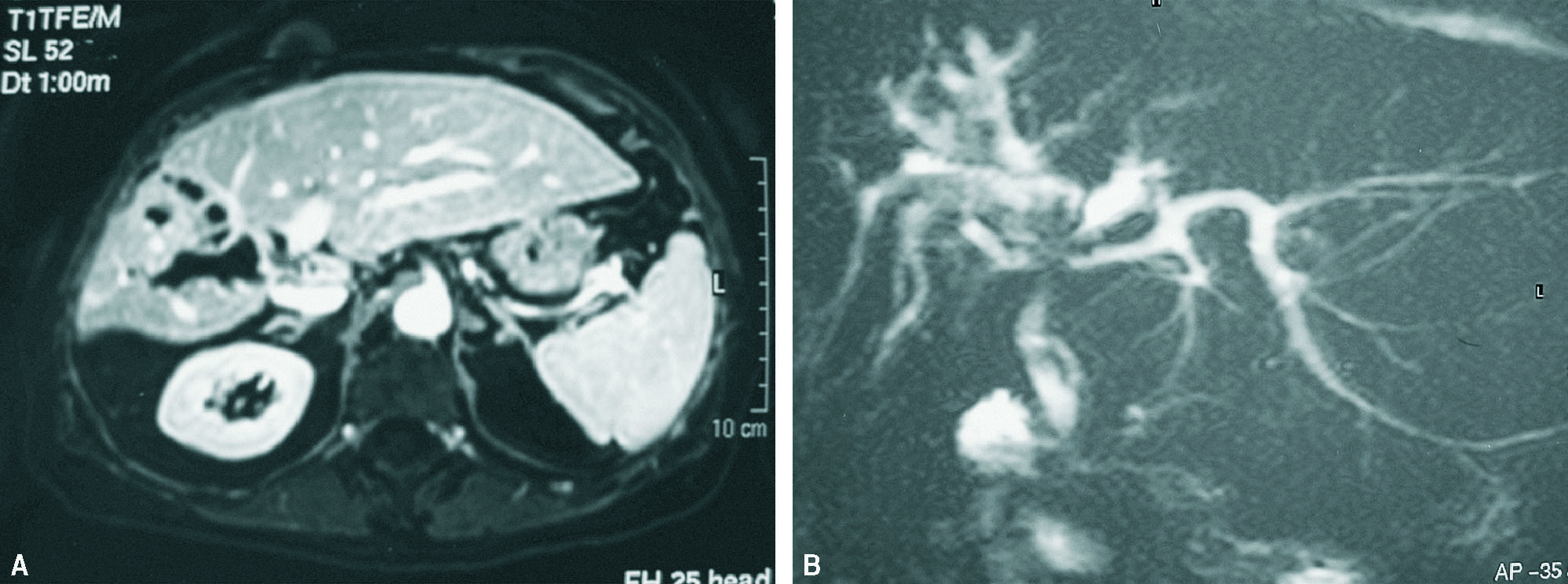
Fig. 3. Resonancia magnética y colangiorresonancia magnética con atrofia del lóbulo hepático izquierdo con dialatación sacular de la vía biliar intrahepática derecha
Discusión
La EC, descrita a principios de siglo y posteriormente caracterizada en 1958 por Caroli, es una rara malformación congénita de la vía biliar intrahepática, formando parte de las malformaciones de la placa ductal que aparecen en el desarrollo del árbol biliar durante el período embrionario. Clasificada según Todani como tipo V en la definición de los quistes de colédoco, se caracteriza por dilataciones saculares, segmentarias, múltiples, quísticas y no obstructivas de los conductos biliares intrahepáticos, conservando la histología normal del parénquima hepático. La forma no hereditaria (tipo I), como los casos que presentamos, es también conocida como "tipo puro esporádico" o enfermedad de Caroli, y por lo general se limita a un lóbulo hepático (más frecuentemente el izquierdo) y se asocia a la formación de litiasis intrahepáticas y colangitis bacteriana recurrente. La forma hereditaria recesiva (tipo II) afecta al hígado de forma difusa y cursa con fibrosis hepática congénita e hipertensión portal secundaria (síndrome de Caroli). Es más frecuente en las mujeres que en los varones, aunque su incidencia exacta en la población general se desconoce, dada la rareza en su diagnóstico.
Las manifestaciones clínicas son variables, y pueden permanecer asintomáticos durante largos períodos y ser un hallazgo casual durante el estudio de otras afecciones, como durante los estudios de insuficiencia renal de nuestro paciente.
La forma de presentación más frecuente se caracteriza por episodios de fiebre por crisis colangíticas secundarias al estasis biliar que producen las dilataciones saculares. En la mayoría de los casos, el primer episodio de colangitis bacteriana ocurre sin evidencia de ningún factor precipitante o tras una exploración de la vía biliar por otros motivos. Se caracteriza por fiebre, sin dolor abdominal y sin ictericia, mínima elevación de la fosfatasa alcalina (FA) y la gammaglutamiltranspeptidasa (GGT), al igual que los estudios analíticos de urgencias que motivaron el ingreso de nuestros pacientes. En la forma compleja predominan las manifestaciones de la hipertensión portal que pueden incluso preceder a las manifestaciones biliares. Las complicaciones más habituales son la cirrosis biliar secundaria, los abscesos intrahepáticos y los tumores; el colangiocarcinoma es una complicación maligna descrita en un 7-24% de los casos1.
La ecografía abdominal es hoy en día el medio de elección para la valoración inicial del paciente con sospecha de EC, incluso es posible su uso en el diagnóstico prenatal2. La colangiopancreatografía retrógrada endoscópica (ERCP) y la colangiografía transhepática percutánea (PTC) se consideran los dos métodos más sensibles para la identificación y caracterización de la anatomía del árbol biliar, con la subsiguiente demostración de la comunicación de las lesiones quísticas con la vía biliar intrahepática y un diagnóstico preciso. Sin embargo, ambas pruebas son invasivas y contaminan la vía biliar, con el riesgo de desencadenar episodios de colangitis, por lo que son descartadas como exploraciones habituales. Actualmente se considera que el procedimiento ideal para el diagnóstico preciso de la EC es la colangiorresonancia magnética (CRMN) con gadolinio, pues aporta valiosa información anatómica sobre el parénquima y la vía biliar, no es invasivo ni ionizante, y es útil para plantear la estrategia quirúrgica. La TC helicoidal también es útil en la valoración de quistes intrahepáticos, aunque rara vez puede confirmar el origen biliar de éstos.
La EC presenta una asociación demostrada a otras afecciones, probablemente en relación con su origen embrionario y sus características hereditarias. Los quistes coledocales y la enfermedad quística renal (enfermedad poliquística renal autosómica recesiva [EPRAR] y dominante [EPRAD], riñón en esponja o enfermedad quística medular) se ven con mayor frecuencia asociadas a la forma compleja o tipo II (síndrome de Caroli: EC con fibrosis hepática), y la asociación de EPRAR y síndrome de Caroli se produce en un 60% según Waters et al3. La mutación de PKHD1, gen asociado a la EPRAR, se ha identificado en pacientes con síndrome de Caroli, aunque el número de casos causados por esta mutación se desconoce2.
La poliquistosis renal del adulto (EPRAD) se ha asociado típicamente a la enfermedad poliquística hepática del adulto y sólo rara vez se asocia a la EC. En la literatura mundial sólo hay descritos unos 30 casos similares a nuestro paciente, en los que se asocia la EC monolobar o difusa y la poliquistosis renal del adulto4-6.
El tratamiento de la EC viene dado por su presentación anatómica y la severidad y frecuencia de los episodios de colangitis, desde el uso de ácido urodesoxicólico, la profilaxis antibiótica continua y seguimiento para las formas más leves, aunque con pobres resultados3. La lobectomía o el trasplante hepático son las opciones terapéuticas para las formas más complicadas, según el grado anatómico de afección y su asociación a fibrosis portal y/o hipertensión portal7-9. El drenaje de la vía biliar con prótesis biliares y/o esfinterotomías por ERCP ha sido descartado, ya que favorece de forma importante los episodios de colangitis bacterianas recurrentes. Este procedimiento sólo debe ser considerado como un tratamiento intermedio ante una futura actitud quirúrgica. El tratamiento considerado como óptimo para las formas focales de la EC, siempre que sea factible, es la hepatectomía parcial4,9,10. En algunos casos en los que se asocia fibrosis portal o afección difusa con episodios recurrentes de colangitis, se ha planteado el trasplante hepático, con buenos resultados7-9. Vandakethu et al4 exponen el caso de una paciente de 25 años diagnosticada de EPRAR, trasplantada renal a la que se le práctica un trasplante hepático, con buenos resultados dada la severidad de los episodios de colangitis en el marco de la inmunosupresión y su amplia expectativa de vida.
Consideramos nuestro caso 1, de avanzada edad, inmunodeficiente con alto riesgo de episodios recurrentes de colangitis severa en la evolución de la ECF como no candidato a trasplante hepático, pero sí candidato a hepatectomía parcial del segmento afecto como única opción terapéutica potencialmente curable, dada la severidad de los cuadros de colangitis en el contexto de la inmunosupresión por su trasplante renal. El paciente presentó un excelente curso postoperatorio durante los siguientes 90 días. Posteriormente presentó una complicación fatal en relación con su estado de inmunodepresión asociada a una complicación séptica tardía de la intervención.
La selección correcta de pacientes candidatos a cirugía y el momento idóneo garantizan el éxito del tratamiento, hecho que se evidencia con la excelente evolución a largo plazo de nuestros casos 2 y 3, de mediana edad, sin comorbilidades, para los que la resección lobar hepática ha sido un tratamiento definitivo, tal y como se ha demostrado en la literatura mundial4,7-10.
El seguimiento de por vida de estos pacientes es obligatorio a fin de diagnosticar precozmente una degeneración neoplásica en el hígado residual afecto.
Correspondencia:
Dr. R. Medrano-Caviedes.
Hospital de la Santa Creu i Sant Pau.
Sant Antoni Maria Claret, 167. 08025 Barcelona. España.
Correo electrónico: rmedrano@santpau.es
Manuscrito recibido el 14-2-2006 y aceptado el 10-1-2007.

