




Introducción
El seudotumor hemofílico es una rara complicación que se presenta en formas graves de hemofilia. La cirugía es el tratamiento de elección y debe llevarse a cabo bajo la supervisión del hematólogo para manejar la infusión apropiada de factor. Presentamos un caso de seudotumor ilíaco tratado mediante cirugía, previa embolización arterial.
Caso clínico
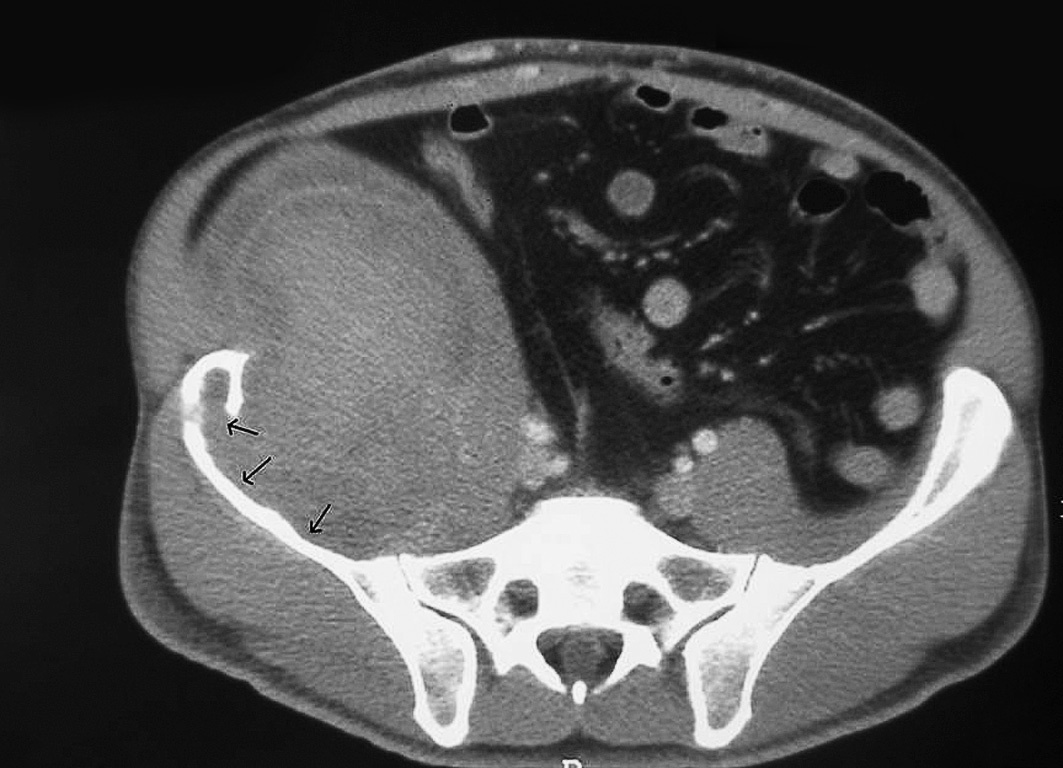
Presentamos el caso de un paciente de 37 años, afectado de hemofilia B, con concentración de factor IX del 2%, virus de la inmunodeficiencia humana en tratamiento con triple terapia, con carga viral inferior a 50, y positivo para virus de la hepatitis C, en tratamiento con interferón. En junio de 2004 ingresó en otro centro por dolor y tumefacción en la cadera derecha. Se le practicó ecografía y se le diagnosticó de hematoma del psoas derecho. La tomografía computarizada mostraba una masa de 17 × 12 × 15 que erosionaba la cresta ilíaca (fig. 1). Los exámenes de laboratorio mostraron un hematocrito del 32%; plaquetas, 166.000; tiempo de Quick, 100%; tiempo de cefalina, 43/30 min, con funciones renal y hepática normales. Se inició tratamiento con factor IX plasmático diario. Se lo derivó a la unidad de hemofilia de nuestro centro tras realización de resonancia magnética en noviembre de 2004. Tras objetivar crecimiento tumoral en dicha exploración, se practicó embolización arterial. En la tomografía computarizada de control a los 3 meses, se visualizó una discreta reducción tumoral, por lo que se decidió nuevo seguimiento radiológico en 3 meses. En la nueva tomografía de agosto de 2005, el tumor había aumentado de tamaño y presentaba mayor destrucción cortical, por lo que se comentó al servicio de cirugía general para plantear abordaje quirúrgico. Se decidió embolizar el territorio ilíaco-glúteo 2 semanas antes de la cirugía, para reducir el tamaño tumoral. Se intervino en septiembre de 2005 procediendo a la resección del seudotumor mediante un abordaje extraperitoneal por una incisión en la fosa ilíaca derecha, con desbridamiento posterior de la cortical interna del hueso ilíaco y pulverización de sellante de fibrina. El postoperatorio cursó sin incidencias y fue alta al decimotercer día para proseguir la rehabilitación funcional.
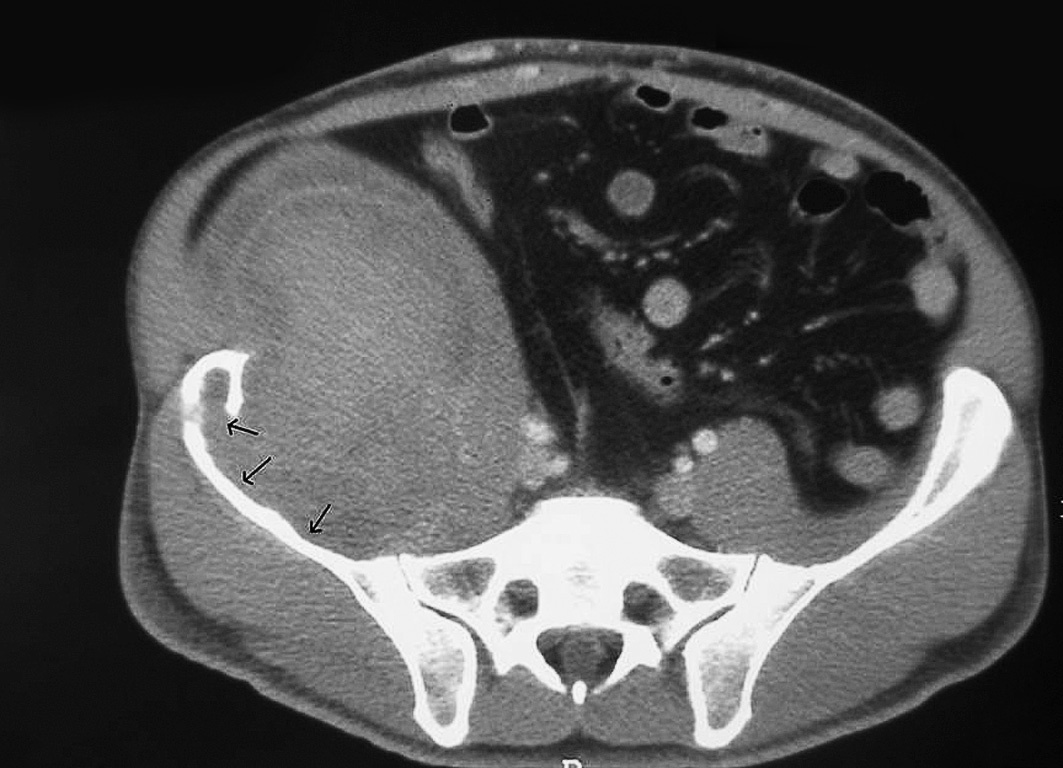
Fig. 1. Tomografía computarizada: seudotumor hemofílico pelviano. Las flechas muestran la destrucción de la cortical del hueso ilíaco.
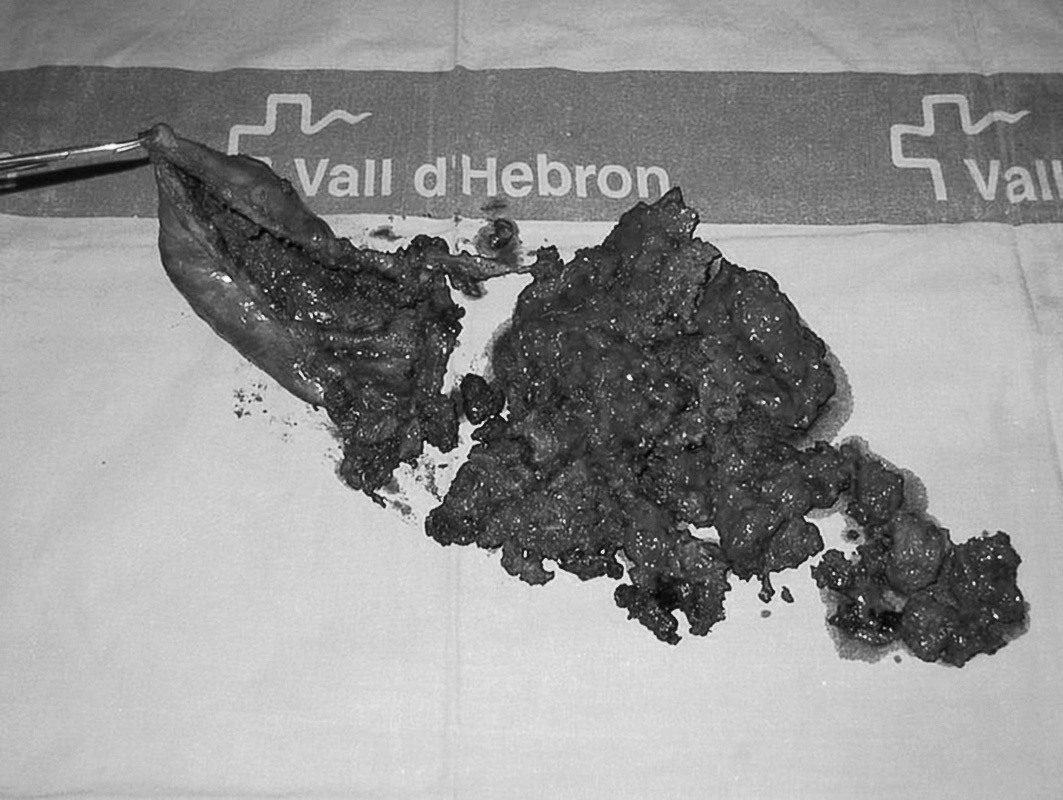
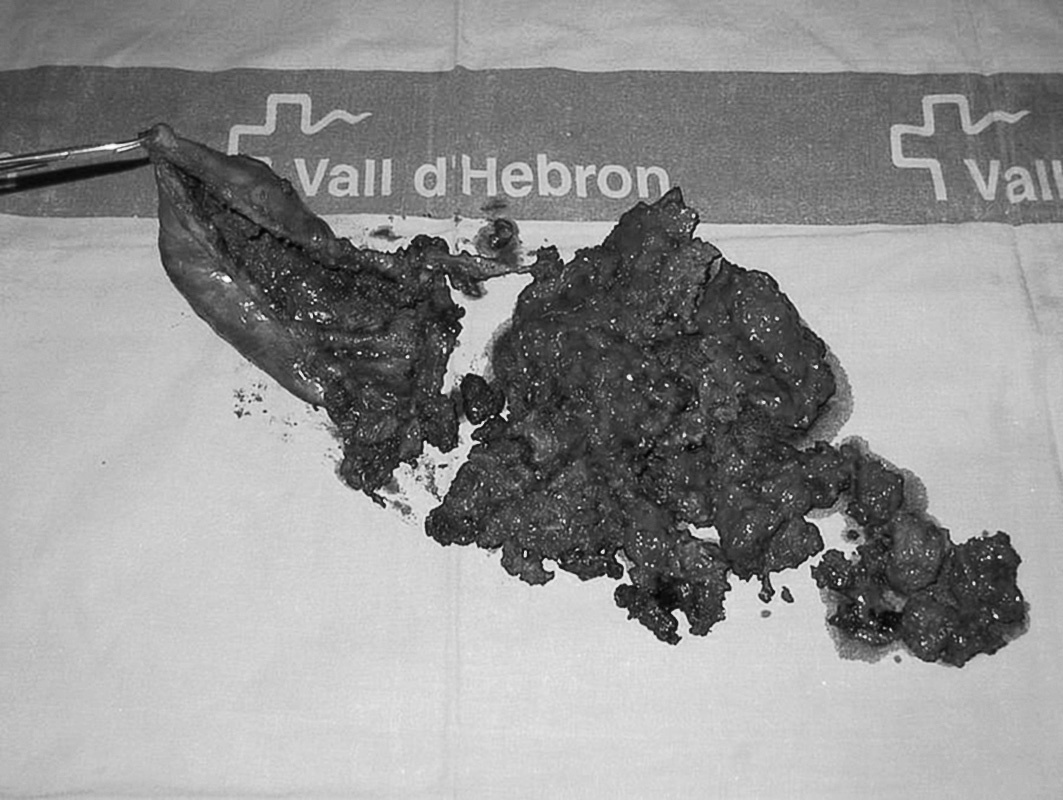
Fig. 2. Pieza quirúrgica: cápsula del seudotumor con contenido hemático interior.
Discusión
Las hemofilias A y B son coagulopatías congénitas caracterizadas por la producción deficitaria del factor VIII (hemofilia A) o IX (hemofilia B), proteínas que actúan como cofactores en la secuencia enzimática de la generación de trombina. Los genes anormales en la hemofilia son transmitidos por el cromosoma X con carácter recesivo. Clínicamente se manifiestan en forma de hemorragias espontáneas repetidas y persistentes, cuya gravedad está en relación con la concentración sanguínea del factor. A diferencia de otras coagulopatías congénitas, hay una predilección por hemorragias articulares y musculares, y es menos frecuente en órganos y sistemas. La manifestación más frecuente es en forma de hemartros. Más del 10% de las complicaciones hemorrágicas en la hemofília ocurren en el músculo1.
El seudotumor hemofílico es una rara y grave complicación de los hematomas musculares típicos de esta enfermedad. Se manifiesta como una masa quística indolora, de crecimiento lento e irregular, que puede comprimir órganos vitales o abrirse al exterior. Es una complicación infrecuente que aparece en un 1-2% de los pacientes con formas severas2. Habitualmente hay un antecedente traumático. Generalmente se localizan en tejidos blandos como el músculo, pero ocasionalmente afectan a los huesos, de los que los más afectados son el fémur, la pelvis, la tibia y los huesos de la mano. Probablemente, el músculo más afectado es el iliopsoas1. Localizaciones descritas más infrecuentes son la órbita, la mandíbula, el maxilar, la clavícula, el cráneo y el canal espinal3-5.
Un seudotumor consiste en productos sanguíneos en diferentes estadios de evolución envueltos por una cápsula fibrosa que contiene macrófagos cargados de hemosiderina. El progresivo crecimiento de estas masas benignas comprime las estructuras adyacentes y causa la destrucción ósea y la necrosis de músculos y piel. Habitualmente se trata de masas indoloras. Las complicaciones son en forma de sobreinfección, formación de fístulas o rotura espontánea con hemorragia en casos raros, aunque usualmente cursan de forma asintomática durante largos períodos.
Gilbert diferenció dos patrones de presentación y comportamiento en función de la localización1. Así pues, los proximales se localizan en el esqueleto axial (fémur, pelvis), son de evolución lenta, aparecen en adultos y no responden al tratamiento conservador. Probablemente se inician como hemorragias de tejidos blandos que erosionan posteriormente el hueso. En contraposición, los distales afectan a individuos jóvenes y se localizan distales al tobillo y la muñeca (calcáneo, metatarso). Tienen un rápido desarrollo y parecen ser secundarios a hemorragias intraóseas.
En cuanto al manejo diagnóstico, la tomografía computarizada es particularmente útil en la evaluación ósea, mientras que la resonancia magnética es superior para determinar la afección de tejidos blandos y espacios intramedulares6.
Hay múltiples alternativas terapéuticas para esta entidad: manejo conservador, exéresis quirúrgica, manejo percutáneo, irradiación y embolización. El manejo de estos pacientes es complejo y debe ser llevado por equipos multidisciplinarios en centros de referencia de hemofilia. Es importante el diagnóstico temprano y la clave para reducir su incidencia es la prevención de los hematomas musculares.
El tratamiento conservador consiste en la infusión de factor VIII y la inmovilización. En estos casos puede haber regresión, aunque no cura. Este tipo de tratamiento se recomienda sólo en pacientes con altos títulos de inhibidores de factor en los que la cirugía no sea posible. En estos casos también puede realizarse la evacuación percutánea con relleno de la cavidad con sellante de fibrina, matriz ósea o hidroxiapatita7.
La escisión quirúrgica es el tratamiento de elección según se establece en la mayoría de las publicaciones. Debe realizarse en centros de referencia porque no está exenta de complicaciones y presenta una tasa de mortalidad del 20%. Se debe realizar minuciosas determinaciones de las concentraciones de factor durante el acto quirúrgico y el postoperatorio. Para rellenar el defecto se han descrito numerosos procedimientos, como la plastia con epiplón pediculizado o con flaps musculares de glúteo mediano o recto del abdomen1,8. Las complicaciones postoperatorias más temidas son la infección, la fistulización y las fracturas patológicas, que en ocasiones requieren la amputación de la extremidad afectada. La embolización arterial es un arma terapéutica que permite reducir la vascularización del tumor, con lo que se reduce el tamaño y, por consiguiente, el riesgo de sangrado durante la cirugía9. La radioterapia con dosis de 10-20 Gy ha sido utilizada en casos en los que la cirugía no es posible y el paciente no responde al tratamiento conservador.
Sin tratamiento, la evolución natural de estos tumores es hacia la destrucción tisular adyacente, con erosión ósea, compresión visceral, neural o vascular10.
Correspondencia:
Dra. S. Castro Boix.
Servicio de Cirugía General. Unidad Esofagogástrica. Hospital Vall d'Hebron.
Pg. Vall d'Hebron, 119-129. 08035 Barcelona. España.
Correo electrónico: sandracastro73@hotmail.com
Manuscrito recibido el 30-11-2005 y acepado el 3-1-2006.

