




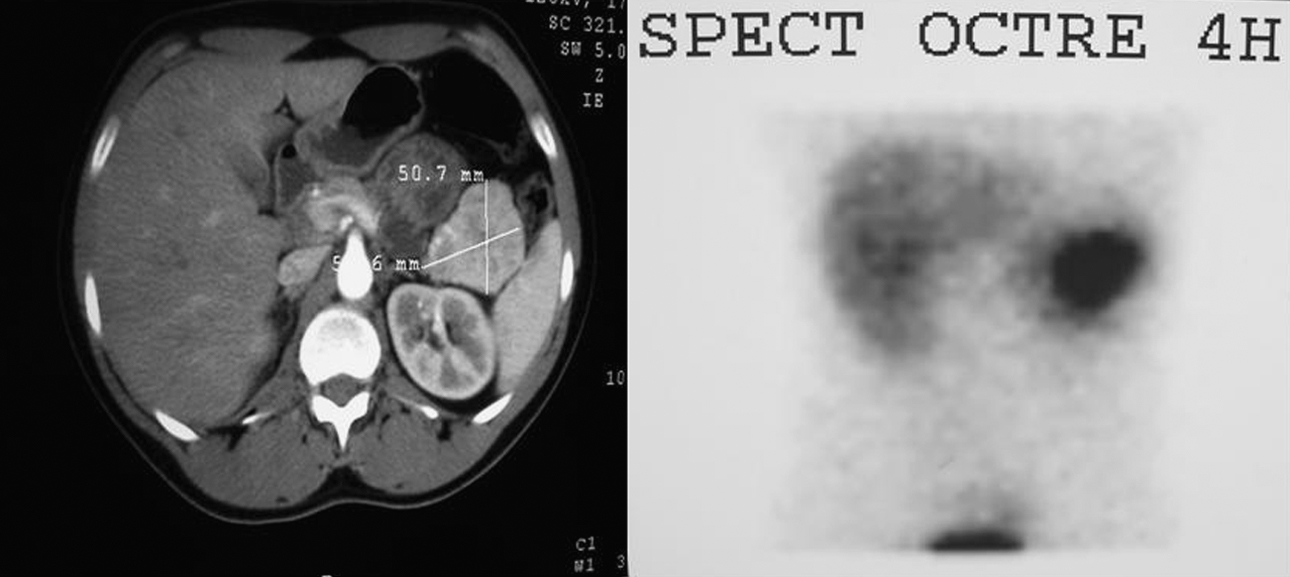
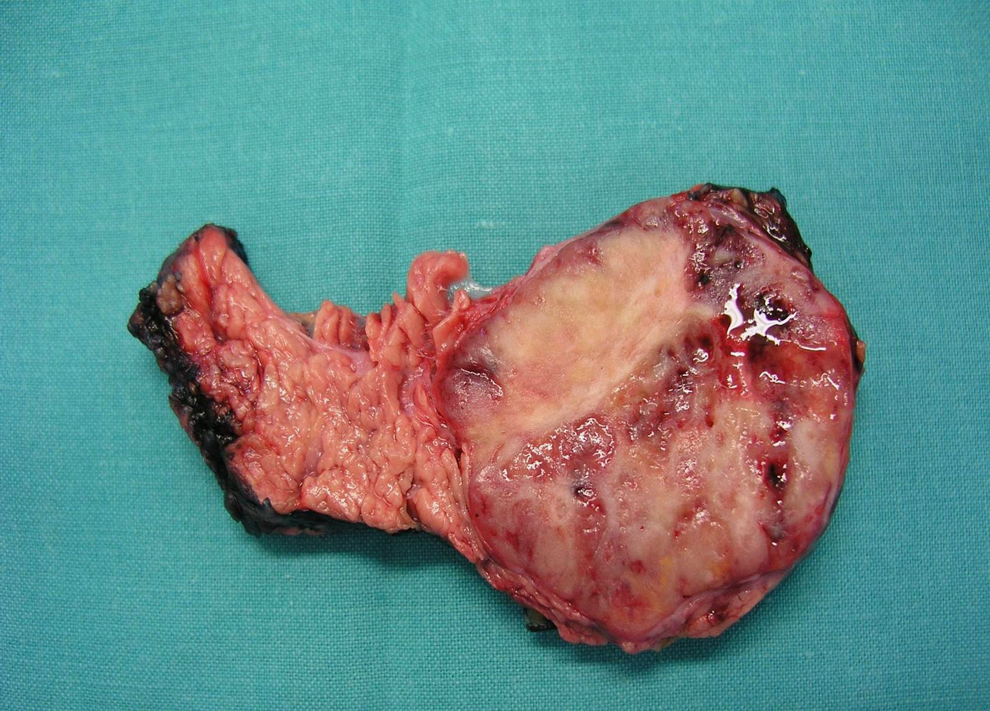
La detección de una concentración elevada de polipéptidos (PP) en sangre apunta a un tumor neuroendocrino no funcionante o un tumor productor de otras hormonas gastroenteropancreáticas. El tumor productor de polipéptido pancreático (PPoma), o tumor de células F, es poco frecuente, generalmente no funcionante y, en ocasiones, es un hallazgo incidental en un estudio de imagen. El carácter asintomático del PP hace posible que el tumor productor pueda alcanzar un gran tamaño y se diagnostique con metástasis a distancia, sobre todo hepáticas1,2,3,4. Es raro que el PPoma produzca síntomas clínicos1,2,3,4,5, aunque en ocasiones conlleva diarrea, úlceras gastroduodenales múltiples, hemorragia digestiva alta o rabdomiólisis por hipopotasemia secundaria a diarrea acuosa1,2,3,4,5,6,7. La literatura recoge tan sólo unos pocos tumores productores exclusivos de PP1,2,3,4, la mayoría, esporádicos y sólo 6 diagnosticados en el seno de un síndrome de neoplasia endocrina múltiple tipo 1 (MEN 1)2,5. Presentamos a un paciente con síndrome de MEN I y con tumor productor exclusivo de PP (PPoma puro). Se trata de un varón de 22 años, asintomático, miembro de una familia portadora del gen del MEN 1, en concreto la mutación InsA 1166 en el exon 8. Durante el programa de detección tumoral, se realiza un estudio bioquímico y hormonal completo para descartar tumores de hipófisis, hiperparatiroidismo y neoplasias duodenopancreáticas o suprarrenales. La única alteración relevante es la elevación del PP sérico de 2.725 (normal < 100)μmol/l. La tomografía computarizada (TC) y la gammagrafía con octreotida revelan una masa de 5cm de diámetro en la cola de páncreas con intenso realce y aumento de vasos en periferia, sin infiltración de estructuras vecinas, adenopatías o metástasis hepáticas (figura 1). Se lo interviene quirúrgicamente, con el hallazgo de una lesión única en la cola del páncreas en íntimo contacto con el bazo, sin adenopatías ni metástasis hepáticas. Se practica una pancreatectomía distal y esplenectomía. El postoperatorio cursa sin complicaciones. No se evidencia recidiva de la enfermedad durante el seguimiento. En el estudio histológico macroscópico se observa un tumor de 4,5cm en la cola del páncreas, bien delimitado, con superficie de aspecto blanco-grisáceo con focos hemorrágicos, con un patrón endocrino bien diferenciado y con ganglios linfáticos aislados negativos (figura 2). El estudio inmunohistoquímico muestra positividad citoplasmática para marcadores endocrinos (sinaptofisina y cromogranina) y en el 90% de las células, para polipéptido pancreático, y negatividad para insulina, glucagón, gastrina, VIP, somatostatina y ACTH, con Ki-67 y actividad proliferativa del 1,6%, por lo que se trata de un PPoma puro.
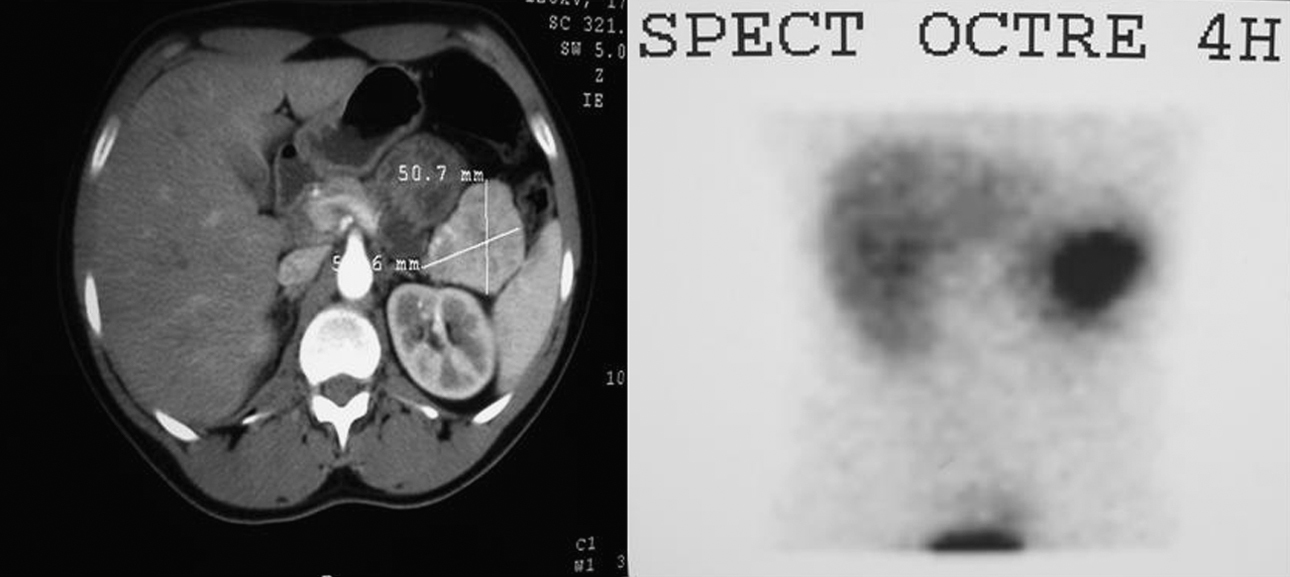
Figura 1. Imagen de tomografía computarizada y gammagrafía con octreotida que muestran una masa en cola de páncreas de unos 5cm.
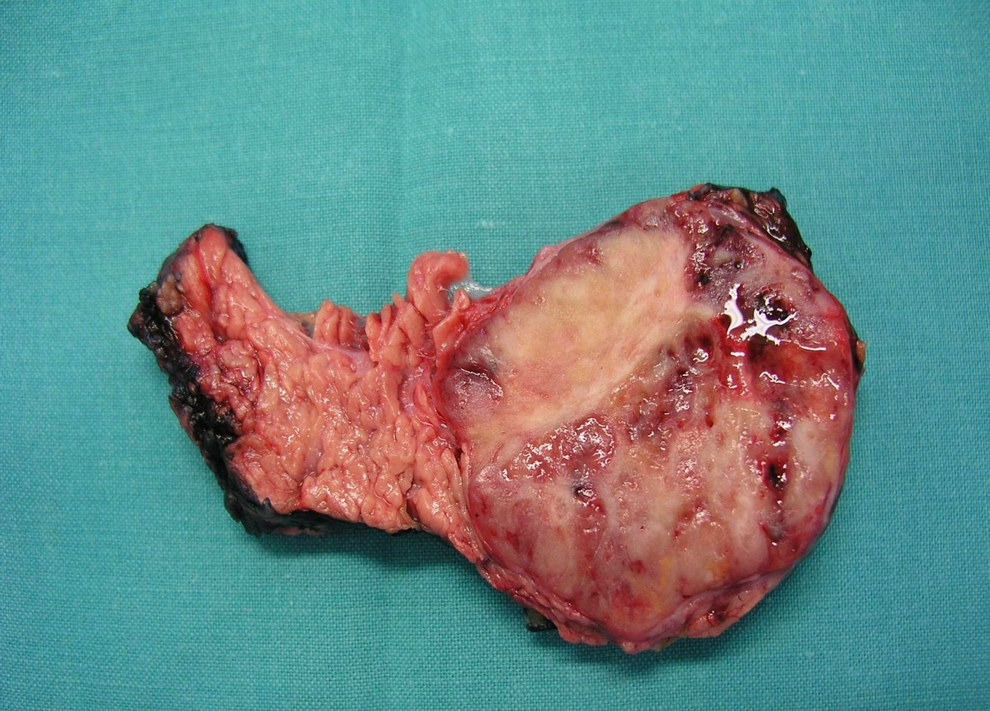
Figura 2. Imagen macroscópica de tumoración en cola de páncreas, bien delimitada y patrón endocrino bien diferenciado.
La detección elevada de PP en plasma sirve como marcador de un tumor neuroendocrino de páncreas, aunque también de otras localizaciones (hígado, papila de Vater, duodeno o estómago)1,2,3. Los PPoma puros son tumores compuestos exclusivamente por células F productoras de PP, y cursan con elevación de PP en plasma. La confirmación diagnóstica requiere estudio inmunohistoquímico positivo exclusivo para PP1,2,3,4. La cromogranina A plasmática también puede estar elevada en los PPoma (sensibilidad del 60–90%). Nuestro caso presentaba valores altos de cromogranina A y PP.
El diagnóstico radiológico se basa en TC o resonancia magnética, con sensibilidad diagnóstica similar. La gammagrafía con octreotida es la técnica no invasiva con mayor sensibilidad en el diagnóstico de tumores neuroendocrinos (mayor del 86% en todas las series), así como en la detección de metástasis2,8. La ecoendoscopia es la prueba de imagen más sensible ante tumores de pequeño tamaño, sobre todo ante aquellos no funcionantes9.
Los pacientes portadores de mutaciones de MEN 1 requieren un seguimiento exhaustivo, con determinaciones bioquímicas y hormonales anuales y pruebas de imagen cada 3–5 años durante toda la vida, desde los 5 años de edad, circunstancia que favorece el diagnóstico precoz durante el seguimiento de la enfermedad de base5,8.
El tratamiento del PPoma supone resección del tumor y linfadenectomía regional2,3,4,5,6,7,8. Los tumores neuroendocrinos tienen una rica vascularización y no suelen necrosarse. Sin embargo, aunque infrecuente, algunos tumores neuroendocrinos presentan un componente quístico central, secundario a necrosis, que puede dificultar el diagnóstico diferencial con lesiones benignas de páncreas y llevar a un diagnóstico preoperatorio erróneo10.
En los casos muy avanzados se puede utilizar quimioterapia, interferón recombinante o análogos de la somatostatina2,8. Asimismo, es necesario un seguimiento frecuente por la posible aparición de metástasis, recidiva local u otros tumores multicéntricos no detectados en el primer acto operatorio. La supervivencia de los pacientes con tumores neuroendocrinos dependerá del tamaño del tumor, las metástasis en el momento del diagnóstico y la concomitancia o no de un síndrome hereditario2,3,4,5,6,7,8.

