




Idiopathic fibrosing pancreatitis (IFP) is a rare form of chronic pancreatitis, mostly occurring in children and adolescents.1 The mean age at diagnosis is around 10 years of age (range 4 months–20 years), and is equally common in both sexes.1,2 It was first described by Comfort et al. in 1946,3 and clinical signs are usually an obstructive jaundice associated with abdominal pain and slight or no elevation of pancreatic enzymes in most cases. It usually generates glandular fibrosis producing a mixed pancreatic insufficiency. Only 72 cases have been described so far in the literature.1,2,4–14
Most cases are diagnosed during exploratory laparotomy and pancreatic biopsy, and in many of these cases a bilio-digestive bypass is performed.2 In a few cases a pancreatic resection has been performed, with or without duodenal preservation.1 In the last few years, several cases of conservative treatment have been reported with favourable results, which has generated a change in perspective with regard to treatment.2,10,12,13
We present a case of IFP that presented in the emergency department with abdominal pain and jaundice and was treated with the suspicion of acute cholecystitis and possible cholangitis. The intraoperative findings and subsequent clinical course made a change in diagnosis necessary after several tests and re-operation. A review of the literature has also been performed.
A 20-year-old man with a prior history of epilepsy and mental handicap came to the emergency department due to acute abdominal pain of 24h duration located in the right upper quadrant and obstructive jaundice. On arrival, the patient was hemodynamically stable and without fever. Physical examination revealed mucocutaneous jaundice and pain located in the right upper quadrant without peritoneal irritation. Blood tests revealed an important elevation of conjugated bilirubin (total bilirubin=91umol/L, alkaline phosphatase=422U/L, GGT=369U/L) as well as an elevation of transaminases (AST=126UI/L, ALT=299UI/L) and a slight elevation of CRP (20mg/L). All other blood test values were normal.
An abdominal ultrasound revealed a distended gallbladder with thickened and edematous walls (4mm), without clear signs of cholelitiasis and dilatation, of the intra and extrehepatic bile duct with a choledocal diameter of 10mm. The pancreatic area was not visible because of gas interposition.
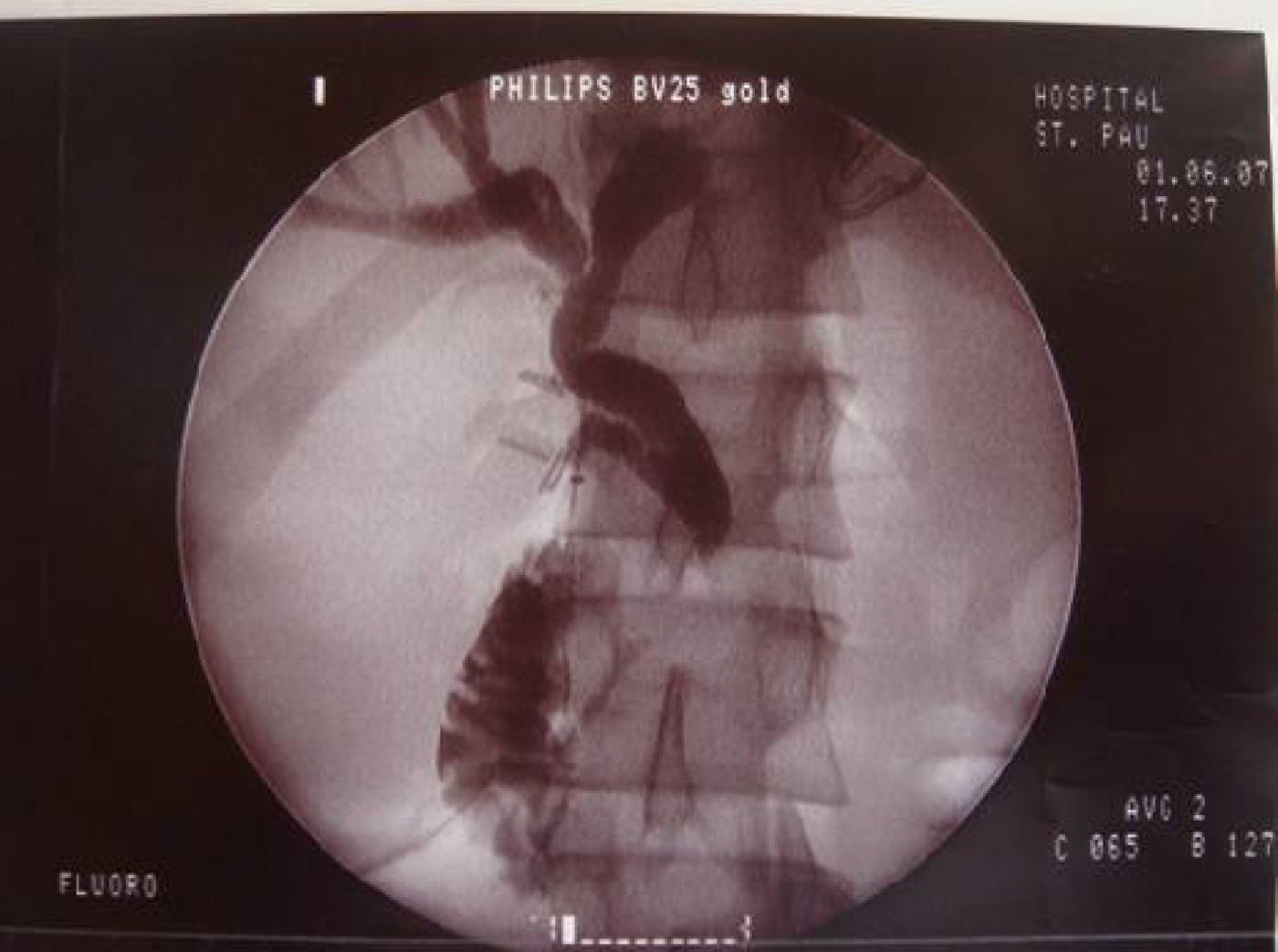
The diagnostic suspicion was of acute cholecystitis with probable cholangitis. A laparoscopic surgical exploration was decided and intraoperatively an edematous and distended gallbladder was found. A transcystic cholangiography was performed that confirmed a diffuse dilatation of the biliary tree with an irregular stenosis and filiform passage of the contrast to the duodenum, without visualization of choledocolitiasis (Fig. 1). An attempt was made to dilate the stenotic area with a Fogarty catheter but it was not achieved. A bile sample was negative for tumoral cells. A cholecystectomy was performed and a Kehr drain was placed in the common bile duct. Endoscopic retrograde cholangiography was not available intraoperatively and was therefore planned for the postoperative period.
 with dilatation of the proximal bile duct and filiform passage of the contrast to the duodenum (white arrow).")
In the postoperative period a high biliary output through the drain was observed (>500–600mL daily). On the 4th postoperative day an ERCP was performed that visualized a normal looking papilla and second portion of the duodenum. However, it was not possible to cannulate the papilla and biopsies were performed extracting samples of the duodenal content for cytology, which were negative for tumoral cells.
Tumoral markers (CEA, CA 19.9 and alfa-fetoprotein) and immunological studies (protein electrophoresis, total immunoglobulins and fractions including IgG4, and anti-carbonic anhydrase) were within normal values.
The study was completed with imaging tests (CT scan, MRI) that showed a global increase in the size of the pancreas without revealing tumoral lesions and an irregular stenosis of the intrapancreatic choledocal duct and dilatation of the rest of the bile duct.
Differential diagnosis was considered between intrinsical inflammatory causes of biliary stenosis (sclerosing cholangitis, primary biliary cirrhosis) or extrinsical causes (chronic pancreatitis) with a possibility of a primary biliary or pancreatic neoplasia.
Because no definitive diagnosis had been made and there was persistent biliary obstruction another surgery was performed. A right subcostal incision was performed that revealed a globally augmented pancreas with a hard consistency. Multiple biopsies were taken, the Kehr tube was removed and biopsies of the stenotic choledocal segment were performed as well as of retroportal lymph nodes. All preoperative biopsies were negative for tumour. Because of the patients age and the low probability of malignancy a biliodigestive derivation was performed: a suprapancreatic section of the common bile duct with closure of the distal end and a termino-lateral Roux en Y hepaticojejunostomy were performed.
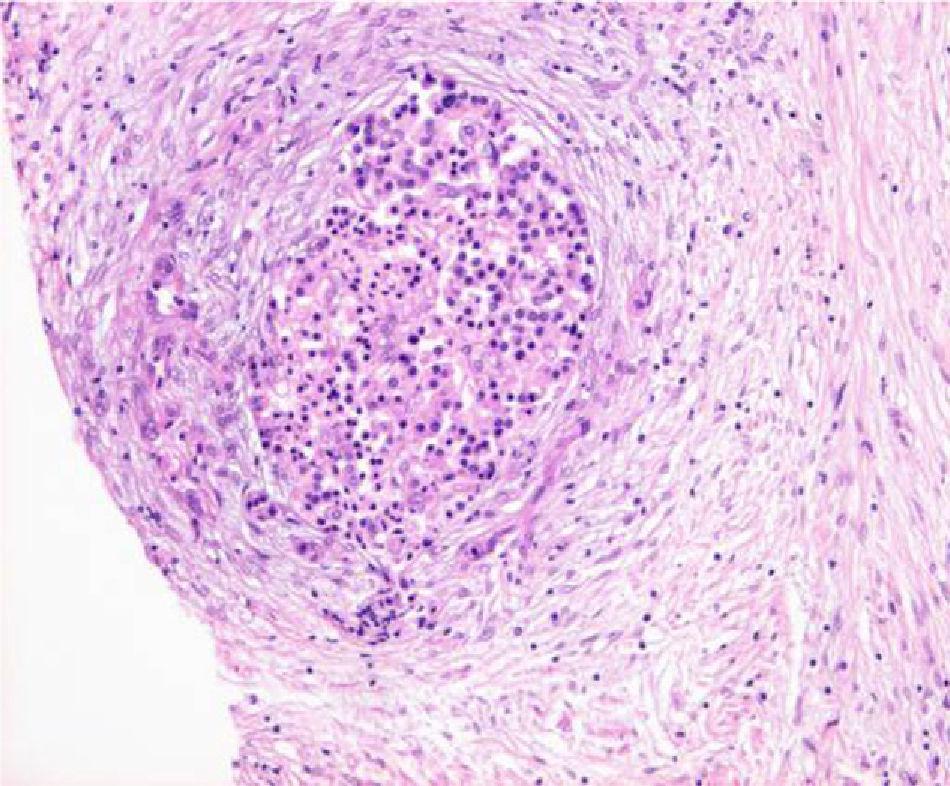
The pathology revealed a severe atrophy of the exocrine pancreatic parenchyma, with fibrosis and chronic inflammation with preservation of Langerhans islets (Fig. 2). These findings, combined with the age of the patient and clinical presentation as an obstructive jaundice syndrome suggested the diagnosis of idiopathic fibrosing pancreatitis. An autoimmune pancreatitis was discarded due to a lack of characteristic pathological findings (lymphoplasmocytic infiltrates, periphlebitis, periductal inflammation) and an absence of suggestive blood tests (increase of IgG4, and anticarbonic anhydrase).15
 – atrophy of the exocrine pancreatic parenchyma with fibrosis (arrow) and preservation of Langerhans islets.")
The postoperative course was uneventful and the patient was discharged on the 10th postoperative day, and remains asymptomatic during follow-up (three years) without need for any further medical or surgical treatment.
Obstructive jaundice in children and adolescents can be caused by a variety of diseases, including congenital biliopancreatic abnormalities, inflammatory processes and tumours. Juvenile chronic pancreatitis is uncommon and has two distinct clinical presentations1:
- (1)
Calcifying pancreatitis, with two variants–a hereditary chronic pancreatitis with autosomal dominant inheritance and tropical juvenile chronic pancreatitis.
- (2)
Non-calcifying or obstructive pancreatitis, which is less common, related to congenital or acquired alterations of the pancreatic ductal system, characterized by a less severe injury of the pancreatic ducts and the absence of pancreatic lithiasis.
Idiopathic fibrosis pancreatitis is a variant of this second type. It is characterized by an extensive fibrosis of the exocrine pancreatic parenchyma with a relative preservation of Langerhans islets. There are several theories on its pathogenesis, including autoimmune reactions related to inflammatory bowel disease or viral infections (parvovirus).16 Recent studies have shown its association with certain genetic mutations (PRSS 1, SPINK 1, CTFR-5 T).17 Due to its rarity and clinical presentation–diffuse or focal pancreatic involvement–the diagnosis is by exclusion, and a histological confirmation is necessary.
In most cases, the most common clinical sign is progressive obstructive jaundice, often accompanied by abdominal pain. Pancreatic enzymes are usually normal or slightly elevated. Imaging studies (ultrasound, CT, MRI) show dilatation of the bile ducts and a pancreas globally increased in size. A magnetic resonance cholangio-pancreatography is considered the diagnostic test of choice by some authors,9 because it has high resolution in showing the duct system and can exclude biliopancreatic anatomical anomalies.
Pathological diagnosis is based on a surgical or percutaneously obtained biopsy (ultrasound, CT). Among mini-invasive options, a fine-needle aspiration biopsy guide by echoendoscopy has recently been used with good efficiency, and can avoid the use of ionizing radiation in paediatric patients.13
Diagnostic confirmation has been obtained in the majority of published cases by surgery, which allows treatment at the same time.1,2,4,6,7,9,16,18,19
Intraoperative examination reveals a globally augmented pancreas, or hard consistency, or the presence of tumoral masses in the pancreatic head. Directed biopsies in the tumoral cases or random biopsies in the diffuse cases are performed. After confirmation of benign disease, the operation usually ends with the creation of a biliodigestive derivation.7,8 Other more aggressive surgical manoeuvres have also been described such as a cephalic duodenopancreatectomy or cephalic pancreatectomy with duodenal preservation, especially in focally involved cases.1 In some cases surgery was followed by an internal biliary drainage by sphincterotomy or sphincteroplasty20 while others only performed an abdominal examination and biopsies.2,12
Finally, ERCP with the placement of a biliary prosthesis has been used successfully in patients with IFP, beginning with the first series published by Sylvester in 1998. Symptom resolution was obtained in all cases, and they were removed some months later.9 Other authors have used endoscopic drainage as a bridge to surgery.1
Other alternatives to surgical or endoscopic treatment have been reported in the literature with favourable response after the use of steroids or conservative measures. Recently, resolution of the jaundice after the administration of ursodoxycholic acid has been described, arguing its capacity to reduce the viscosity of bile with a subsequent decrease in the obstruction.10,12 In these cases, resolution of symptoms was observed in approximately 2 months. Authors such as Harper et al.10 consider this as a reasonable period of time, before performing an endoscopic or surgical derivation. Globally, conservative management avoided surgical biliary derivation in a high percentage of cases.2,9,10,12 Subsequently, none of these patients had a relapse after the initial presentation.9 However, the progression of fibrosis and subsequent atrophy of the pancreatic parenchyma has been described in the majority of cases, and in some cases an exocrine pancreatic insufficiency has appeared, and less frequently endocrine insufficiency.1,9,10
According to the knowledge on the natural evolution of IFP and taking into consideration the therapeutical advances of recent years, we propose a staged approach to this disease. In a paediatric/adolescent patient with obstructive jaundice, IFP is an exclusion diagnosis that requires histological confirmation. Once the diagnosis has been confirmed, management should be performed by a multidisciplinary team including paediatrician, gastroenterologist, radiologist and surgeon. The first stage would be an observation period because there have been cases described of spontaneous resolution.2 If the obstructive jaundice persists or worsens a second stage can be proposed using medical treatment with steroids or ursodeoxycholic acid that could be used for a period of approximately 2 months according to the previously mentioned references.10 Finally, when resolution has not been achieved an endoscopic or surgical biliary derivation should be proposed.
Please cite this article as: Mocanu SN, Artigas Raventós V, Rodríguez Blanco M, Farré Viladrich A, Trias Folch M. La pancreatitis idiopática fibrosante: causa infrecuente de ictericia obstructiva en pacientes jóvenes. Cir Esp. 2013;94:271–274.
