




Although strategies have been defined for the management of certain inherited cancers (breast, ovarian, colorectal, and endometrial), the management of patients with mutations involving increased risk of cancer is a major concern that has not been defined.
Germline mutations in the TP53 gene result in a rare inherited condition known as Li-Fraumeni syndrome (LFS).1
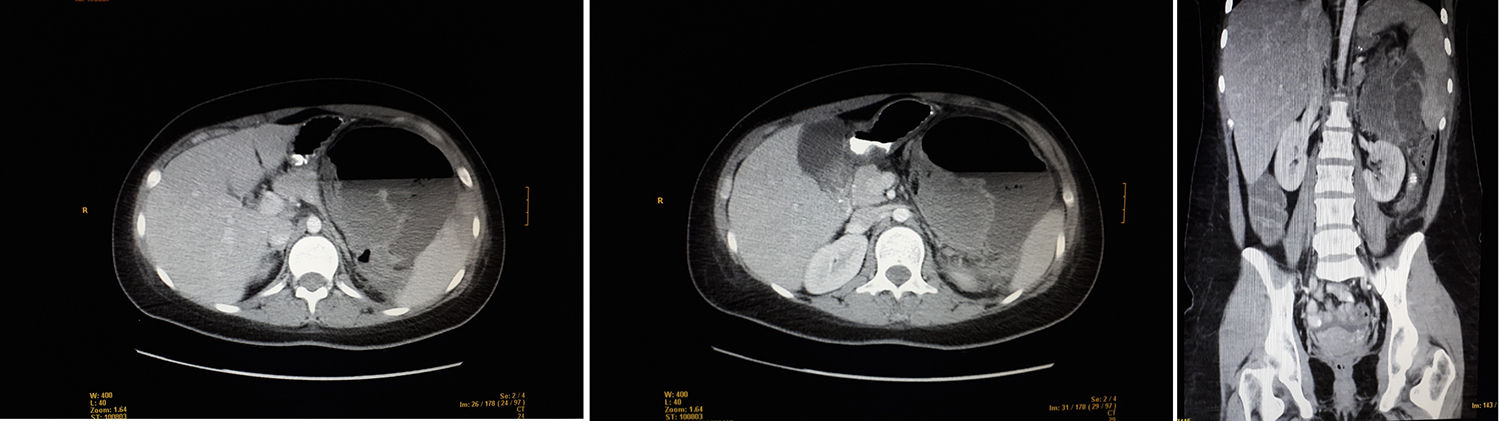
We present the case of a 21-year-old woman with abdominal pain in the left hypochondrium and fever. At the age of 11, she had been diagnosed with osteosarcoma of the right proximal humerus, with no metastasis. She had been treated with the SEOP 2001 chemotherapy protocol (ifosfamide days 1–5 and adriamycin 25mg/m2 days 1–3) before and after surgery with right interscapulothoracic disarticulation. The patient had residual neuroma, which was followed-up annually until July 2017. Family history included a maternal grandfather who had died from lung cancer (age 69) and paternal grandfather from prostate cancer (age 78). Physical examination revealed a distended, painful abdomen in the left hypochondrium and a mass, but no abdominal guarding. Lab work showed leukocytosis, neutrophilia and CRP 340. CT scan detected a heterogeneous mass measuring 10×5cm in the left hypochondrium, which was extensively necrotic and abscessed, displacing neighboring structures and presenting air-fluid level. Other observations included: splenomegaly with areas suggestive of infarctions, liver with no focal lesions, kidneys without findings, no pathological lymphadenopathies, and moderate intraperitoneal free fluid.
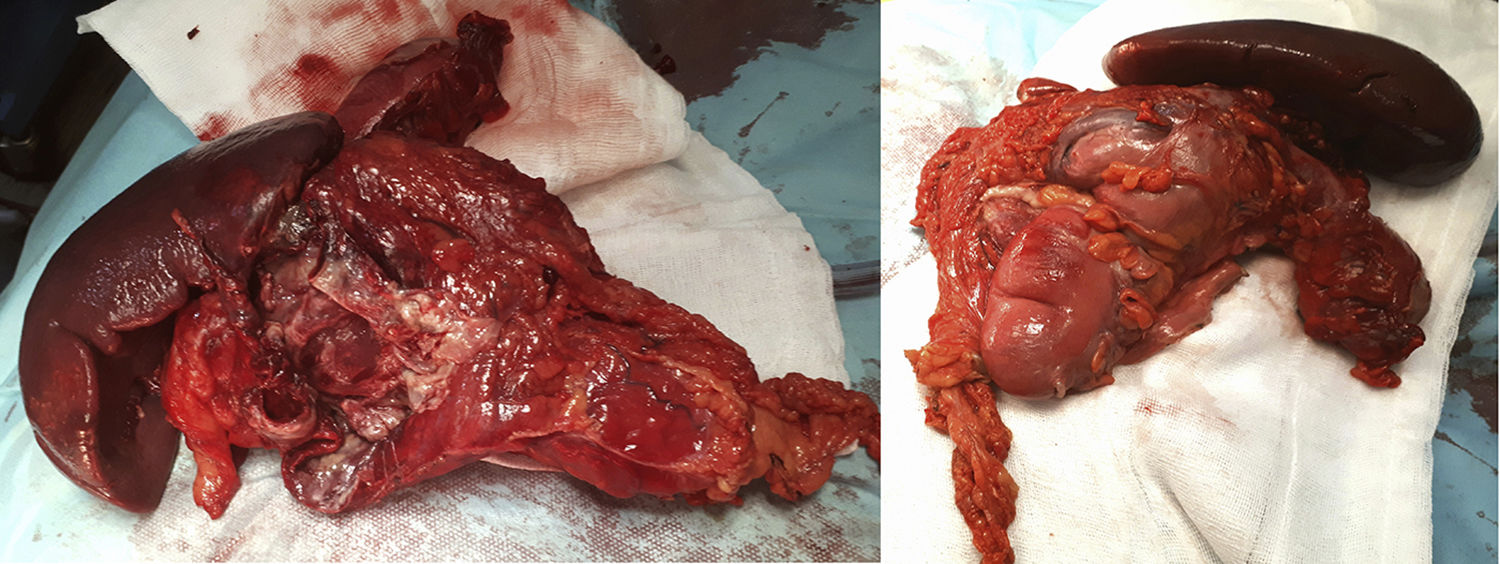
The surgery identified a purulent peritonitis of 2.5L with a dependent mass of the distal transverse colon embedded anterior to the splenic hilum. At a posterior level, it was in contact with the tail of the pancreas and the greater curvature of the stomach up to the fundus. No space-occupying lesions were palpated in the liver. After left hemicolectomy and splenectomy with primary reconstruction of the intestinal tract, the patient recovered the transit on the fifth postoperative day and was discharged after extension studies with brain and breast MRI, as well as a genetic study at the National Oncology Research Center (Fig. 1).

The pathological anatomy study identified a gastrointestinal stromal tumor (GIST) that was multifocal, low-grade and fusiform, measuring 5×3cm, with free surgical margins and no lymphovascular or perineural invasion. There were less than 50 mitoses per high-power field (G1), and no evidence of necrosis was observed. Peritoneal cytology was negative for malignancy. Immunohistochemical panel showed: CD117 positive (++); Dog1 focal positivity; CKAE1/AE3 negative; S100 negative (−); CD34 positive (+); Ki-67 proliferative in approximately 10%–15% cellularity; and actin negative (−) (Fig. 2).

A history of pediatric cancer should lead us to the hypothesis of a hereditary cancer predisposition syndrome, especially with a family history or concurrent congenital abnormalities. Conditions associated with increased risk of childhood cancer include: genodermatosis (neurofibromatosis type 1 and tuberous sclerosis), overgrowth syndromes (Beckwith-Wiedemann syndrome and Proteus syndrome) and adult cancer syndromes with an increased risk of childhood and adolescent cancer (LFS and Li-Fraumeni-like syndrome (LFLS).2
LFS is a cancer predisposition syndrome with autosomal dominant inheritance. Patients with LFS are at increased risk of developing multiple primary tumors, with an 83% risk of having a second primary if the first neoplasm was diagnosed between 0 and 19 years.3
There are 2 forms of presentation: LFS and SFLL.4 The classic definition of LFS requires a diagnosis of sarcoma before age 45, together with a first-degree relative with any cancer before age 45, as well as a first or second-degree relative with any cancer before age 45 or a sarcoma diagnosed at any age.5
The inclusion criteria have been modified in the last 30 years by Birch, Eeles 1 and 2, and Chompret in 2001. In 2009, the Chompret criteria were redefined: patient with LFS spectrum cancer (sarcoma, breast, central nervous system tumor, leukemia, bronchoalveolar carcinoma) before the age of 46 and a first or second-degree relative with an LFS spectrum tumor (except previous breast cancer) before the age of 56 or multiple tumors. It is also defined as a patient with multiple tumors, at least 2 LFS spectrum, the first occurring before age 46, or an adrenocortical carcinoma or choroid plexus carcinoma at any age or breast cancer before age 36 without BRCA1 or BRCA2 mutation.1
In 1990, inactive forms of the TP53 gene were observed in sporadic forms of many tumors associated with LFS, thereby discovering a definitive association between TP53 and LFS.6
In germ cells, there are more than 250 mutations in the TP53 gene.7 A TP53 gene mutation can be identified in patients with no family history because of insufficient family history, germline mosaicism or a de novo mutation of the TP53 gene.8
Clinical criteria for LFS/LFSL can estimate the probability of germ cell mutation in the TP53 gene: 70% of families with the classic criteria of LFS, 32% Chompret criteria, 25% modified Chompret, 25% Birch criteria, 14% Eeles 1 and 8% Eeles 2 criteria.9,10
The development of new clinical markers under study in carriers of TP53 germline mutations (decrease in telomere length), as well as the increasingly common use of multiple genetic tests, will continue to expand the new phenotypes beyond the classic description of LFS in the future.7
Please cite this article as: Ruiz Gómez F, de Miguel Ibañez R, Moreno Serrano A, Pérez Dominguez T, Palomo Sánchez JC. Síndrome de Li-Fraumeni. Cir Esp. 2019;97:600–602.
