




Descrito por Wermer1, el síndrome de neoplasia endocrina múltiple tipo 1 (MEN 1) es un trastorno con una prevalencia de 2/100.000 habitantes. Se caracteriza por la asociación de adenomas paratiroideos, tumores neuroendocrinos gastroenteropáticos y adenomas hipofisarios2, con predisposición a otras lesiones como tumores adrenales, carcinoides, angiofibromas faciales y meningiomas entre otros3.
Por su parte, los tumores neuroendocrinos pancreáticos (TNEP) representan el 3% de las lesiones en dicha localización. La mayoría son esporádicos, pero pueden aparecer en el seno de endocrinopatías, como el MEN 1.
Mujer, 55 años, con diagnóstico genético de MEN 1 tras ser intervenida a los 17 años por microprolactinoma y a los 26 por hiperparatiroidismo primario en relación a hiperplasia paratiroidea.
Durante el seguimiento se realizan determinaciones hormonales (calcio, PTH, glucosa, insulina, glucagón, IGF-1, prolactina y cromogranina A) anuales y ecografía, tanto cervical como abdominal cada 3 años sin hallazgos durante 20 años.
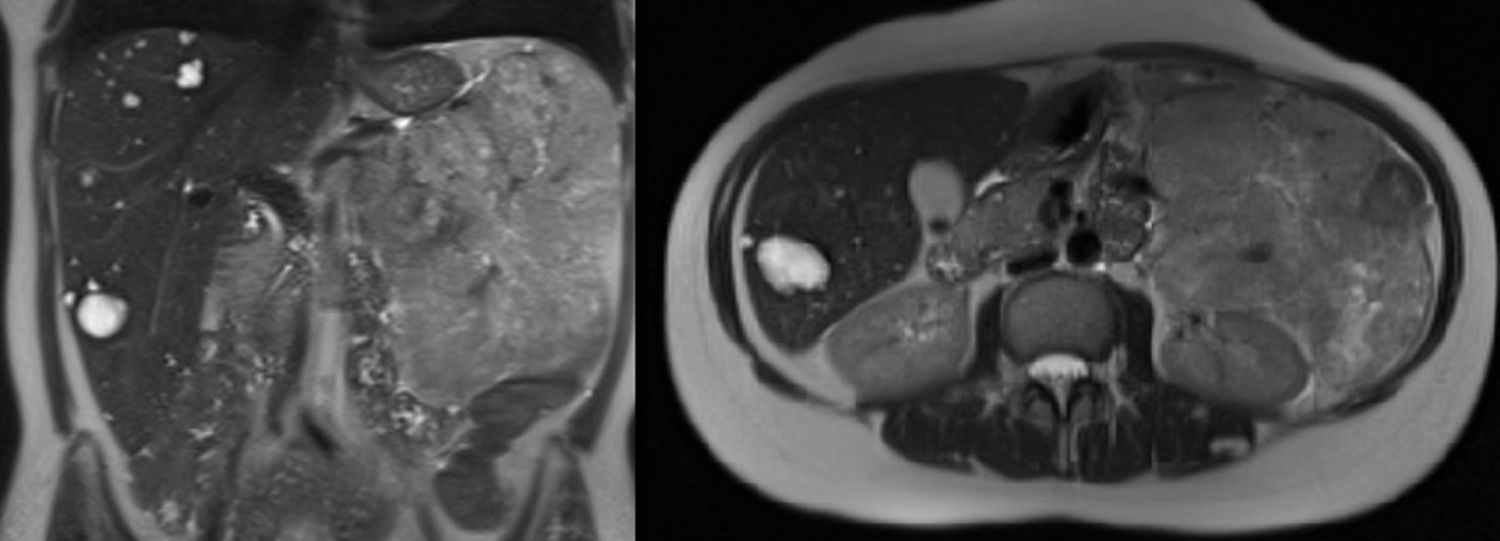
Tras hallazgos no concluyentes en última ecografía, se realiza resonancia magnética (RM) en el siguiente control, donde se objetiva tumoración retroperitoneal de 16×13×10cm en contacto con cuerpo-cola pancreáticas y riñón izquierdo (fig. 1).

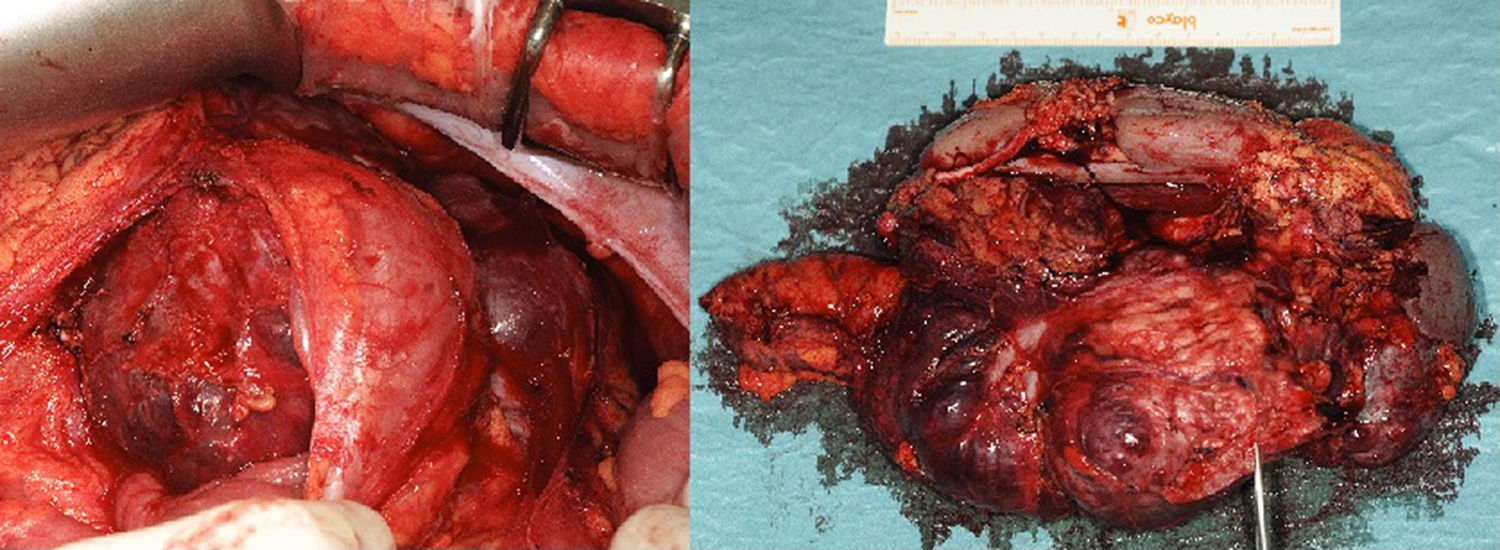
Con diagnóstico de sospecha de TNEP no funcionante se decide intervención quirúrgica, realizando resección en bloque, incluyendo cola de páncreas, bazo, riñón izquierdo y colon descendente por infiltración (fig. 2).

El resultado histopatológico muestra un tumor neuroendocrino (22×16×7cm) moderadamente diferenciado de origen pancreático, que sobrepasa los límites pancreáticos (pT3), con márgenes de resección libres.
A los 12 meses la paciente no ha recibido tratamiento adyuvante y está libre de enfermedad (estudio con octreotide sin evidencia de lesiones).
El MEN 1 es un trastorno de herencia autosómica dominante, aunque un 10% se asocia con mutaciones de novo3. Su alteración genética más frecuente se encuentra en el gen MEN 1 (cromosoma 11), con penetrancia de más del 95% a los 40 años4.
Los adenomas paratiroideos son la manifestación más frecuente, con penetrancia de casi el 100% a los 40-50 años, y típicamente la más precoz, con presentación unos 30 años antes que los casos esporádicos2.
Los adenomas hipofisarios presentan una incidencia entre el 15-50%, siendo el prolactinoma el más frecuente. Tienen tendencia a mayor tamaño, mayor agresividad y menor respuesta al tratamiento, pero sin aumentar la prevalencia del carcinoma3.
En cuanto a los TNEP asociados suelen ser de aparición más precoz, múltiples y con propensión a degeneración maligna5.
Tradicionalmente, el gastrinoma con síndrome de Zollinger-Ellison (40%) era el más frecuente6. El incremento de las pruebas de imagen ha hecho que los tumores no funcionantes sean los más frecuentes de la región pancreático-duodenal (30-80%), con una penetrancia del 35% a los 50 años. Más de un 70% no son realmente no funcionantes, sino que segregan sustancias como el polipéptido pancreático, enolasa o neurotensina7, pero sin repercusión clínica.
El hecho de no estar asociados a ningún síndrome clínico y que la sintomatología vaga asociada hace que el diagnóstico sea en estadios avanzados, con un tamaño mayor a 5cm en un 70% y metástasis al diagnóstico en más del 60%7.
Las indicaciones de cirugía vienen dadas por el crecimiento tumoral o el tamaño >2cm, por debajo no hay beneficios en supervivencia8,9. En el manejo de lesiones de 1-2cm, debido a la morbimortalidad asociada a la cirugía pancreática y a la aparición de nuevas lesiones en el remante pancreático3,6, no existe consenso sobre el momento óptimo ni la extensión de la cirugía.
En cuanto al screening de los TNEP asociados a MEN 1 no existe un protocolo estándar, sino que varían, siempre incluyendo screening bioquímico anual (calcio, PTH, prolactina, glucosa, insulina y cromogranina A) y variabilidad en cuanto a las pruebas de imagen. En algunos casos se recomienda ecoendoscopia, TC toraco-abdominal (si antecedente de cirugía pancreática o carcinoide tímico) y gammagrafía de receptores de somatostatina (si TNEP malignos) cada 3 años10; mientras que otros proponen el uso de pruebas de imagen cada 3-5 años2 o anuales3, dependiendo siempre de los deseos de la paciente. En nuestro hospital, actualmente, a raíz de este caso, se realiza determinación bioquímica anual y RM abdominal cada 3 años.
La supervivencia es menor en este grupo poblacional frente a la población similar en edad, sexo y otras características demográficas, con probabilidad de muerte del 50% a los 50 años sin tratamiento. Los TNEP son los principales indicadores pronósticos en MEN 1 y la malignización de estos la principal causa de muerte3. El screening no se ha relacionado con un aumento de la tasa de supervivencia10.
El tamaño mayor a 3cm y la presencia de metástasis están relacionados con el pronóstico, de modo que un diámetro a partir de 1,5-2cm se relaciona con un aumento del riesgo de malignización5,6.
Por tanto, nuestro caso es relevante por la presencia de TNEP no funcionante de gran tamaño sin metástasis al diagnóstico, lo que supone una cierta mejoría en el pronóstico de la paciente, y por plantear la necesidad de estandarizar el seguimiento radiológico en estos pacientes.
FinanciaciónNo ha recibido ninguna beca para su realización.

