




As described by Wermer,1 multiple endocrine neoplasia type 1 (MEN 1) syndrome is a disorder with a prevalence of 2/100000 inhabitants. It is characterised by the association of parathyroid adenomas, gastroenterohepatic neuroendocrine tumours and pituitary adenomas,2 with a predisposition towards other lesions, such as adrenal and carcinoid tumours, facial angiofibromas, meningiomas, etc.3
Pancreatic neuroendocrine tumours (PNET) represent 3% of the lesions in said location. Most are sporadic but can appear in endocrinopathies such as MEN 1.
The patient is a 55-year-old woman with a genetic diagnosis of MEN 1 after having undergone surgery at the age of 17 due to microprolactinoma and at 26 for primary hyperparathyroidism related with parathyroid hyperplasia. Follow-up included annual hormone determinations (calcium, PTH, glucose, insulin, glucagon, IGF-1, prolactin and chromogranin A) and cervical/abdominal ultrasound every 3 years, with no abnormal findings for 20 years.
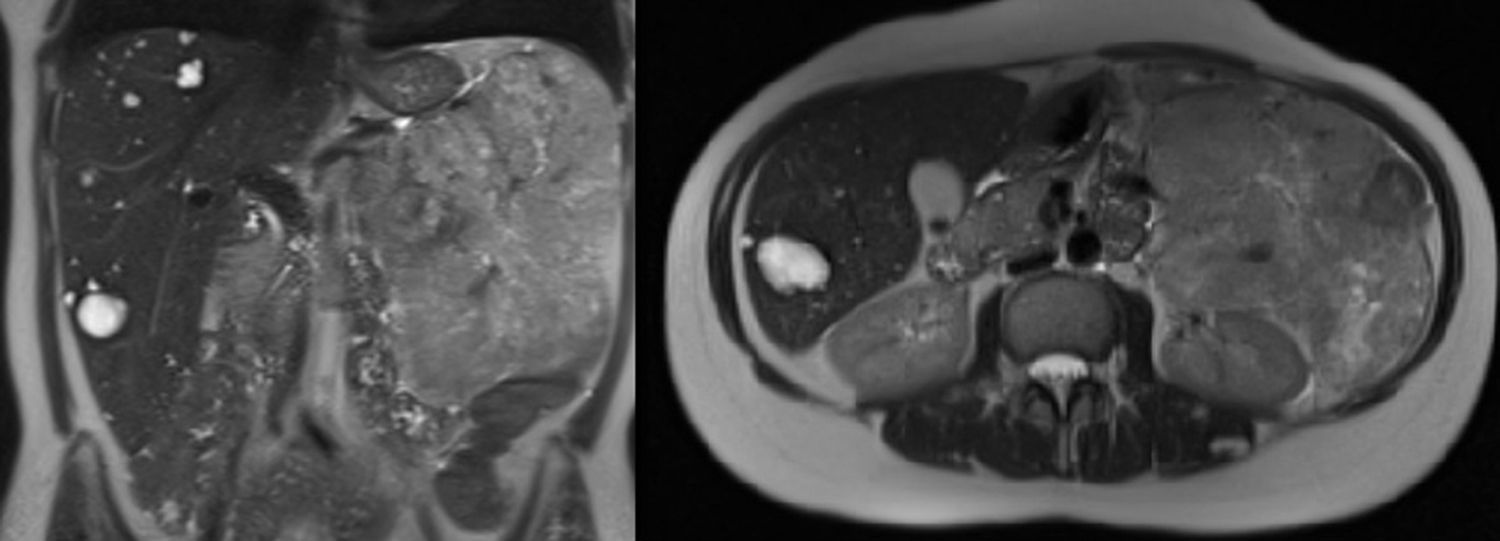
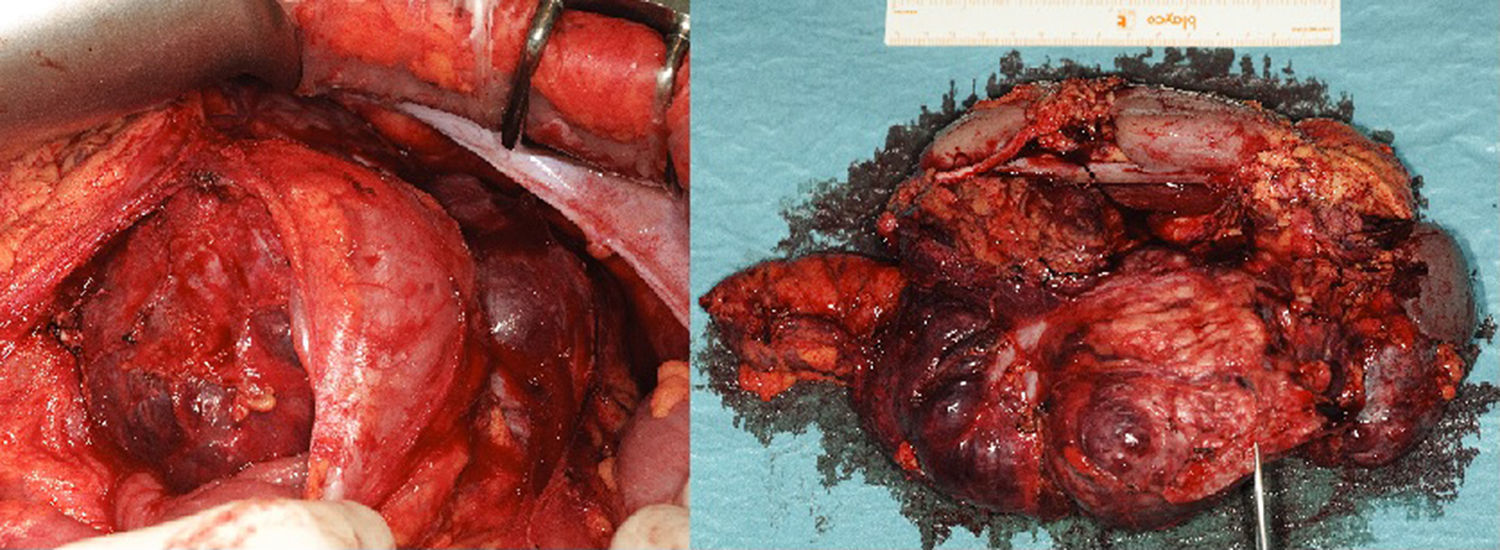
After inconclusive findings on the last ultrasound, magnetic resonance imaging (MRI) was used at the next follow-up, which detected a retroperitoneal mass measuring 16cm×13cm×10cm in contact with the body/tail of the pancreas and left kidney (Fig. 1). With a suspected diagnosis of non-functioning PNET, we decided to operate and performed en bloc resection including the tail of the pancreas, spleen, left kidney and descending colon due to infiltration (Fig. 2). The histopathology study reported a neuroendocrine tumour (22cm×16cm×7cm) that was moderately differentiated and pancreatic in origin, which surpassed the pancreatic limits (pT3); resected margins were disease free.


Twelve months later, the patient has received no adjuvant treatment and is disease free (octreotide study showed no evidence of lesions).
MEN 1 is an autosomal dominant genetic disorder, although 10% are associated with de novo mutations.3 The most frequent genetic alteration is in the MEN 1 gene (chromosome 11), with a penetrance greater than 95% at the age of 40.4
Parathyroid adenomas are the most frequent manifestation, with a penetrance of almost 100% at ages 40–50, and typically the earliest, with a presentation some 30 years before sporadic cases.2
Pituitary adenomas present an incidence of 15%–50%, and prolactinomas are the most frequent. They tend to be larger, more aggressive and less responsive to treatment, although the prevalence of carcinoma does not increase.3
Associated PNET usually appear earlier, are multiple and tend to degenerate towards malignancy.5
Traditionally, gastrinomas with Zollinger–Ellison syndrome were more frequent (40%).6 Advances in imaging studies has resulted in non-functioning tumours being the most frequent in the pancreatic-duodenal region (30%–80%), with a penetrance of 35% at 50 years. More than 70% are not truly non-functioning; instead, they secrete substances like pancreatic polypeptide, enolase or neurotensin,7 but entail no clinical repercussions.
As they are not associated with any clinical syndromes and the associated symptoms are vague, diagnosis is made in advanced stages and at sizes larger than 5cm in 70% of cases; metastasis is found at the time of diagnosis in 60% of patients.7
Indications for surgery are based on tumour growth or size >2cm; at smaller sizes, no benefits are seen in survival.8,9 In the management of lesions measuring 1–2cm, due to the morbidity and mortality associated with pancreatic surgery and the appearance of new lesions in the remaining pancreas,3,6 there is no consensus about the optimal moment for surgery or its extension.
As for the screening of PNET associated with MEN 1, there is no standard protocol, but it should always include an annual biochemical screening (calcium, PTH, prolactin, glucose, insulin and chromogranin A) and variable imaging studies. In some cases, endoscopic ultrasound, thoracoabdominal CT (if there is a history of pancreatic surgery or thymic carcinoid tumour) and somatostatin receptor scintigraphy (if malignant PNET) are recommended every 3 years.10 Meanwhile, others propose the use of imaging studies every 3–5 years2 or annually,3 always depending on the patient's wishes. At our hospital and based on this case, biochemical determinations are currently done annually and abdominal MRI is done every 3 years.
Survival is shorter in this population group compared to a population that is similar in age, sex and other demographic characteristics, with a probability of death of 50% at the age of 50 if not treated. PNET are the main prognostic indicators in MEN 1 and their malignisation is the main cause of death.3 Screening has not been related with increased survival rates.10
Sizes larger than 3cm and the presence of metastasis are related with prognosis, and diameters of 1.5–2cm are related with increased risk of malignisation.5,6
Therefore, our case is relevant due to the presence of a large, non-functioning PNET without metastasis at diagnosis, which resulted in a certain improvement in the prognosis of the patient. We also emphasise the need to standardise the radiological follow-up in these patients.
FundingNo funding was received for this article.
Please cite this article as: Manuel Vázquez A, Sanz Muñoz P, López López B, Carabias Hernández A, Jover Navalón JM. Manejo diagnóstico/terapéutico en los tumores neuroendocrinos pancreáticos asociados a MEN 1. Cir Esp. 2015;93:539–541.
