




Dietas ricas en fructosa producen, en ratas, hipertrigliceridemia y esteatosis hepática, como consecuencia de una reducción en la actividad transcripcional del receptor activado con proliferadores peroxisómicos alfa (PPARα), junto con un estado de resistencia parcial a la leptina. En el presente trabajo, hemos estudiado cómo la fructosa afecta en el ámbito molecular al control del metabolismo energético hepático.
Material y métodosSe distribuyeron de forma aleatorizada a 16 ratas Sprague-Dawley macho en 2 grupos: control y fructosa (10% peso/volumen en agua de bebida, durante 14 días). Adicionalmente, se distribuyó a 12 ratas en 3 grupos: control, fructosa y fructosa + 1 β-D ribofuranósido de 5-aminoimidazol 4-carboxamida (AICAR) (500mg/kg/día, los últimos 3 días de dieta fructosa). Se valoraron los valores plasmáticos de triglicéridos, glucosa, leptina, insulina y adiponectina. En el ámbito hepático se determinaron: contenido de triglicéridos y ceramidas, actividad de β-oxidación, actividad AMPK (AMP activada proteína cinasa), proteína fosfatasa 2A (PP2A) y valores de expresión proteica y de ácido ribonucleico mensajero de diferentes genes implicados en el metabolismo energético.
ResultadosLas ratas del grupo fructosa presentaron hipertrigliceridemia (×1,3), hiperleptinemia (×1,9) e hiperadiponectinemia (×1,7), incremento en los triglicéridos (×1,6) y ceramidas hepáticas y en la formación de complejos PPARα-FoxO1. La administración de AICAR, aunque incrementó en un 30% la actividad AMPK hepática, no revirtió ninguna de las alteraciones descritas. En ratas fructosa no sometidas a ayuno antes del sacrificio, se detectó en hígado un marcado incremento en la subunidad catalítica de PP2A (×1,6) y en la capacidad de unión de ChREBP (×1,4).
ConclusionesEl déficit de oxidación de ácidos grasos producido por el consumo de fructosa en forma líquida no es reversible por activación de la AMPK hepática. La activación de PP2A tras la ingesta de fructosa es la alteración molecular determinante en la aparición de los cambios metabólicos inducidos por este monosacárido.
Administration of fructoseenriched diets to rats induces hypertriglyceridemia and hepatic steatosis as a result of reduced PPARα transcriptional activity and partial leptin resistance. In the present work, we studied how fructose alters the control of hepatic energy metabolism at the molecular level.
Material and methodsSixteen Sprague-Dawley male rats were randomised to 2 treatment groups: control and fructose (10% w/v fructose in drinking water for 14 days). Additionally, 12 rats were distributed into 3 groups: control, fructose, and fructose+AICAR (500mg/kg/day, in the last 3 days of fructose administration). Plasma levels of triglycerides, glucose, leptin, insulin and adiponectin were measured. Triglyceride and ceramide content, fatty acid β-oxidation activity, AMPK and PP2A activity and expression of mRNA and protein levels of genes involved in energy metabolism were determined in liver samples.
ResultsFructose-fed rats showed high plasma concentrations of triglycerides (×1.3), leptin (×1.9) and adiponectin (×1.7), an increase in hepatic triglycerides (×1.6) and ceramides and in the formation of PPARα-FoxO1 complexes. AICAR administration, despite increasing hepatic AMPK activity (×1.3), modified none of the above mentioned alterations. In livers of non-fasted fructose-fed rats, there was a marked increase in the expression of the catalytic subunit of PP2A (×1.6) and in the binding of ChREBP (×1.4).
ConclusionsThe deficit in liver fatty acid oxidation induced by fructose ingestion in liquid form is not reversed by the activation of AMPK. PP2A activation by fructose ingestion is the key molecular event in the production of fructoserelated metabolic changes.
El consumo de fructosa constituye una parte importante de la ingesta calórica total en las dietas occidentales, básicamente debido al aumento del consumo de bebidas refrescantes que contienen valores altos de fructosa. Durante las últimas 2 décadas, el incremento en el consumo de fructosa ha coincidido con un aumento en la prevalencia de obesidad y síndrome metabólico, importantes factores de riesgo para la enfermedad cardiovascular1,2. Ha habido un intenso debate para determinar si la masiva incorporación de fructosa en las dietas occidentales representa sólo otro ingrediente de alto valor calórico o si, además de esto, la fructosa provoca cambios cualitativos en el metabolismo energético que favorecen la aparición de enfermedades metabólicas3,4. Además, la fructosa incluida en la dieta en forma líquida provoca hipertrigliceridemia en un grado mayor que si ésta se administra en forma sólida5. Un aumento del 10% de la energía total obtenida en la dieta en forma de hidratos de carbono no es suficiente para inducir hipertrigliceridemia, a menos que este 10% se consuma en forma líquida y sea rico en monosacáridos6. Además, en una revisión reciente de Bray et al7, los autores explican que en modelos de obesidad y resistencia a la insulina en roedores, el consumo de fructosa, añadida en el agua de bebida, induce a los roedores el deseo de consumir soluciones edulcoradas y calóricas, lo que reduce el consumo de comida sólida, aunque no en un grado suficiente como para prevenir un equilibrio calórico positivo y, por lo tanto, la aparición de obesidad. Respecto a este tema tan importante, todavía falta conocer el mecanismo molecular encargado de las alteraciones metabólicas provocadas por la ingestión de fructosa.
Recientemente, nuestro grupo ha demostrado que la administración de fructosa al 10% en el agua de bebida a ratas macho provoca hiperleptinemia y un estado de resistencia a la leptina, debidas a una disfunción parcial en el factor de transcripción STAT3 (Signal Transducer and Activator of Transcription-3). Las ratas suplementadas con fructosa, aun presentando valores elevados de leptina en plasma, no presentan diferencias en la expresión proteica de STAT3 fosforilada en serina (STAT-PSer727), forma con máxima actividad transcripcional, respecto las ratas control. Esta falta de actividad puede explicar los cambios metabólicos inducidos por la fructosa cuando ésta se toma en forma líquida8. Aunque no hay pruebas experimentales de que este fenómeno también ocurra en humanos, hay estudios que explican que la administración de fructosa al 20% en solución, a varones sanos voluntarios durante 4 semanas, induce, entre otros efectos, un incremento significativo de la concentración plasmática de la leptina9. Igualmente, hemos demostrado que la administración de fructosa en el agua de bebida a ratas macho Sprague–Dawley provoca un estado de resistencia parcial a la leptina como consecuencia de la falta de fosforilación y activación de STAT3-PSer727 y AMPK (AMP activada proteína cinasa). La falta de estas actividades se debe al déficit de activación de Janus activated kinase 2 (JAK-2) y a una disfunción en la activación del receptor de la leptina que tiene como consecuencia la falta de actividad de la vía de las mitogen activated protein kinase (MAPK)10.
En el presente estudio, demostramos que el déficit de oxidación de ácidos grasos producido por el consumo de fructosa en forma líquida no es reversible por activación de la AMPK hepática. Igualmente, aportamos datos experimentales que indican que la activación de la proteína fosfatasa 2A (PP2A) tras la ingesta de fructosa es la alteración molecular determinante en la aparición de los cambios metabólicos inducidos por este monosacárido.
Material y métodosAnimales y diseño experimentalPara este estudio, se utilizaron 16 ratas macho Sprague-Dawley que fueron proporcionadas por Harlan (Barcelona [España]) y se mantuvieron con agua y comida ad libitum, a temperatura y humedad constante con un ciclo de luz/oscuridad de 12h. Después de 5 días, se distribuyó a los animales de forma aleatorizada en 2 grupos: un grupo control y un grupo suplementado con fructosa (fructosa). La fructosa se proporcionó en el agua de bebida en forma de solución al 10% (peso/volumen) durante 2 semanas. Los animales control no recibieron ningún suplemento de hidratos de carbono. Los animales se sacrificaron por decapitación con anestesia por isoflurano entre las 10.00 y la 11.00h, con o sin ayuno de 2h previo al sacrificio.
Para los experimentos con AICAR, los animales se distribuyeron de forma aleatorizada en 3 grupos de 4 animales cada uno: un grupo control, un grupo suplementado con fructosa y un grupo suplementado con fructosa al que se le administró, vía subcutánea, una dosis de 500mg/kg/día de AICAR los últimos 3 días de tratamiento. Las condiciones del estudio fueron las mismas descritas anteriormente. Todo el proceso se realizó de acuerdo con la guía establecida por el Comité de Bioética de la Universitat de Barcelona, según se indica en la Ley (5/1995) (21 de julio) de la Generalitat de Catalunya.
Preparación de muestrasLas muestras sanguíneas se recogieron en el momento del sacrificio de los animales, en tubos con ácido etilendiaminotetraacético (EDTA) al 5%; el plasma se obtuvo por centrifugación y se conservó a −20ºC hasta su utilización. Los hígados de las ratas se extrajeron y perfundieron en cloruro sódico (NaCl) 0,9%; 1g de tejido hepático de cada rata se homogeneizó en tampón 150mM NaCl, 1mM DTT, 30mM EDTA, 50mM KH2PO4, pH 7,4 para obtener la fracción de sobrenadante posnuclear, que se conservó a −80ºC hasta su utilización. Se congelaron 10–100mg de tejido hepático en N2 líquido de forma inmediata y conservados a −80ºC hasta su utilización para la extracción del ácido ribonucleico (ARN) total. Se conservaron 2 muestras adicionales de tejido hepático (250mg) a −80ºC para la cuantificación de los lípidos hepáticos y para la obtención de los extractos nucleares y de proteína total.
Análisis de lípidos, leptina y adiponectinaLos valores plasmáticos de triglicéridos se midieron mediante el ensayo colorimétrico de Wako Chemicals GmbH (Neuss [Alemania]) Triglyceride L-Type kit. Los valores plasmáticos de leptina y adiponectina se determinaron con el kit RL83K RIA y MADP-60HK RIA, respectivamente, ambos de Linco Research (Missouri [Estados Unidos]). Los lípidos hepáticos se extrajeron y midieron según se describe previamente11.
Ensayos enzimáticosβ-oxidación de los ácidos grasos. La actividad hepática de β-oxidación de ácidos grasos se determinó según el método descrito previamente12, utilizando 30μg de sobrenadante posnuclear de hígado de rata como muestra.
5'-AMP-activada proteína cinasa. AMPK se estudió en la fracción 6% de glicol polietileno (PEG) 8000 analizando la incorporación de [γ32P] ATP en el péptido SAMS (Upstate Biotechnology, Lake Placid, NY [Estados Unidos]), tal como describen Kudo et al13. Brevemente, se homogeneizaron 100mg de tejido congelado en 0,4ml de tampón 50mM Tris–HCl (pH 7,5), 0,25mM de manitol, 1mM de ácido etilenglicoltetracético (EGTA), 1mM de EDTA, 1mM de DDT, 50mM de floruro sódico (NaF), 1mM de fenilmetilsulfonil flúor (PMSF), 5mM de ortovanadato y 1mM de benzamidina. El homogenizado se centrifugó a 14.000 xg durante 20min a 4ºC, y el sobrenadante obtenido se llevó hasta un 2,5% (w/v) PEG 8000 utilizando una solución stock 25% (w/v) PEG 8000. La mezcla se mantuvo en agitación durante 10min y se centrifugó a 10.000 x g durante 10 minutos. El sobrenadante se llevó a un 6% PEG 8000. Después de la centrifugación, el pellet se lavó con tampón de homogeneización al 6% de PEG 8000 y posteriormente se resuspendió en tampón 100mM Tris–HCl (pH 7,5), 1mM de EGTA, 1mM de EDTA, 1mM de DDT, 50mM de NaF, 1mM de PMSF, 1mM de benzamidina, 0,02% azida sódica y 10% glicerol. La concentración proteica de cada muestra se estudió mediante el método Bradford14.
La reacción para analizar la actividad AMPK se realizó a 30ºC durante 5min en un volumen final de 25μl compuesto por 40mM de HEPES-NaOH (pH 7,0), 80mM de NaCl, 8% de glicerol, 0,8mM de EDTA, 200μM de péptido SAMS, 0,8mM de DTT, 5mM de cloruro de magnesio (MgCl2), 200μM de [γ32P] ATP, 200μM de 5'-AMP y 0,5μg de la fracción proteica 6% PEG 8000. La reacción se inició con la adición de [γ32P] ATP/ Mg. Después de la incubación a 30ºC, se añadieron 15μl del producto de reacción a filtros de papel Whatman P81 que se lavaron 4 veces con ácido fosfórico 150mM durante 30min. El último lavado se realizó con acetona, los filtros se dejaron secar y se contaron en un contador de centelleo. El blanco de la reacción se realizó utilizando todos los componentes y añadiendo proteína inactivada. La actividad AMPK se calculó como la diferencia entre la radioactividad de las muestras, menos la del blanco. Se han realizado experimentos preliminares para determinar los tiempos óptimos de incubación y para establecer la cantidad de proteína a utilizar.
Determinación del contenido hepático de diacilglicerol y ceramidasEl contenido hepático de ceramidas se determinó mediante el método de la diacilglicerol cinasa15. Los lípidos se extrajeron a partir de 100mg de tejido con 600μl de cloroformo, metanol y HCl 1N (100:100:1). Tras la agitación y la centrifugación posterior, la fracción clorofórmica se evaporó bajo corriente de N2. Los lípidos secos se resuspendieron con 300μl de KOH 0,1N en metanol y se incubaron 1h a 37ºC para eliminar el diacilglicerol. Tras añadir 300μl de PBS, se repitió la extracción. Finalmente, los lípidos se resuspendieron en 100μl de tampón de reacción (150μg/100μl cardiolipina, 280μmol/l de ácido dietilenetriaminepentaacético, 51mmol/l de octil β-D-glucopiranosida, 50mmol/l de NaCl, 51mmol/l de imidazol, 1mmol/l de EDTA, 12,5mmol/l de MgCl2, 2mmol/l de ditiotreitol, 0,7% de glicerol, 70μmol/l de β-mercaptoetanol, 500μmol/l de ATP y 5μCi/100μl (γ-32 P)ATP) y 35ng de la enzima diacilglicerol cinasa. Las muestras se incubaron a 30ºC durante 30min. La reacción se paró con la adición de 170μl de tampón de parada (135mM de NaCl, 1,5mM de CaCl2, 0,5mM de glucosa y 10mM de Hepes pH 7,2) y 30μl de EDTA 100mM. Los lípidos se extrajeron nuevamente con 1ml de cloroformo, metanol, 1N HCl (100:100:1) y tras resuspender los pellets con 50μl de cloroformo, se cargaron en placas de sílica-gel (TCL Plates, Whatman), usando una solución de cloroformo:metanol:ácido acético (65:15:5) como solvente para fluir. Las placas se expusieron a un film fotográfico para interpretar los resultados.
Para la determinación del DAG, se omitió la hidrólisis alcalina de 1h a 37ºC para eliminar el DAG de las muestras y se siguió el mismo protocolo descrito antes.
Preparación y análisis del ARNEl ARN total se aisló mediante el reactivo Trizol® (Invitrogen Biotechnologies), de acuerdo con las instrucciones del fabricante. Los valores relativos de cada ARN específico se determinaron por la reacción de la transcriptasa reversa-reacción en cadena de la polimerasa (RT-PCR) tal como se ha descrito previamente11. Se utilizó aprt (adenosil fosforibosil transferasa) como control interno. Los oligonucleótidos específicos utilizados fueron: aprt, sentido: 5'-AGCTTCCCGGACTTCCCCATC-3', antisentido: 5'-GACCACTTTCTGCCCCGGTTC-3', tamaño amplificado 329bp; L-piruvato cinasa, sentido: 5'-TATGGCGGACACCTTCCTGGA-3', antisentido: 5'-GCTGAGTGGGGAGGTTGCAAA-3', tamaño amplificado 259bp.
Aislamiento de los extractos nucleares y la proteína totalLos extractos nucleares y de proteína total se obtuvieron según el método descrito previamente10. La cuantificación de la concentración proteica de cada fracción se realizó mediante el método de Bradford14.
Ensayos de Western blotSe sometieron 30μg de proteína total (para PP2A) o extractos nucleares hepáticos (para ChREBP) a electroforesis en geles de poliacrilamida-SDS al 10%. Posteriormente, las proteínas se transfirieron a membranas immobilon polifluoruro de vinilideno (PVDF) (Millipore, a Bedford, MA [Estados Unidos]) y se bloquearon durante 1h a temperatura ambiente en solución de TBS-0,1% Tween-20 con un 5% de leche en polvo desnatada. Las membranas se incubaron siguiendo el protocolo descrito anteriormente8. La detección se realizó utilizando el reactivo ECL de quimioluminiscencia kit HRP (Amersham Biosciences). Como control de carga se incubaron las membranas con β-actina o β-tubulina (Sigma-Aldrich). El tamaño de las proteínas se comprobó utilizando marcadores de peso molecular (Invitrogen, Life Technologies). Todos los anticuerpos se obtuvieron de Santa Cruz Biotechnology (Santa Cruz, CA [Estados Unidos]).
CoinmunoprecipitaciónLos ensayos de coinmunoprecipitación se realizaron a partir de 50μg de extractos nucleares, tal como se ha descrito anteriormente16.
Ensayo de retardación de la movilidad electroforéticaPara la determinación de la actividad de unión del ChREBP al ácido desoxirribonucleico (ADN), se utilizó el oligonucleótido siguiente: 5-tcctgcatgtgccacaggcgtgtcacc-3'. El ensayo de retardación de la movilidad electroforética (EMSA) se realizó tal como se describe previamente8. Para los ensayos de superretardación (superShift), los anticuerpos se añadieron 30min antes que la sonda marcada radiactivamente. Todos los anticuerpos utilizados se obtuvieron de Santa Cruz Biotechnology (Santa Cruz, CA [Estados Unidos]).
EstadísticaLos resultados se expresan como la media ± desviación estándar. Las muestras de plasma se estudiaron por duplicado. Las diferencias estadísticas se valoraron mediante la prueba de la t de Student para muestras no apareadas, utilizando el programa informático GraphPad InStat (GraphPad Software V2.03). Cuando el número de muestras era pequeño o las desviaciones no fueron homogéneas, se aplicó un test no paramétrico (Mann–Whitney o Kruskal-Wallis). Se consideró significación estadística un valor de p < 0,05.
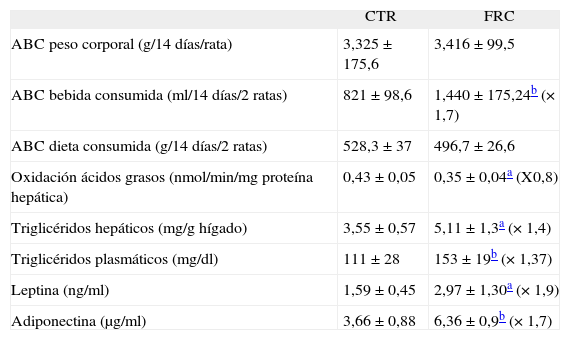
ResultadosLa administración de AICAR revierte el déficit de oxidación de ácidos grasos inducido por fructosaLas ratas suplementadas con fructosa al 10% presentan un incremento en el consumo de agua (×1,7), sin presentar cambios significativos en el peso corporal ni en el consumo de dieta sólida. Las ratas del grupo fructosa son hipertrigliceridémicas (×1,3) y presentan concentraciones plasmáticas elevadas de leptina (×1,9) y adiponectina (×1,7).
Además, las ratas del grupo fructosa presentan unos valores de triglicéridos hepáticos elevados (×1,6) y una reducción en la actividad de β-oxidación de los ácidos grasos (×0,8) (tabla 1). Leptina y adiponectina controlan el catabolismo de los ácidos grasos y la producción de glucosa en el tejido hepático, en parte a través de la activación de AMPK17. Nuestro grupo ha demostrado anteriormente que las ratas del grupo fructosa no presentan cambios en la expresión proteica de la forma fosforilada y activa de AMPK (AMPK-PThr) en el ámbito hepático8, así como en su actividad específica10.
Parámetros analíticos en ratas control (CTR) y ratas suplementadas con fructosa en forma líquida (FRC)
| CTR | FRC | |
| ABC peso corporal (g/14 días/rata) | 3,325 ± 175,6 | 3,416± 99,5 |
| ABC bebida consumida (ml/14 días/2 ratas) | 821 ± 98,6 | 1,440 ± 175,24b (×1,7) |
| ABC dieta consumida (g/14 días/2 ratas) | 528,3 ± 37 | 496,7 ± 26,6 |
| Oxidación ácidos grasos (nmol/min/mg proteína hepática) | 0,43 ± 0,05 | 0,35 ± 0,04a (X0,8) |
| Triglicéridos hepáticos (mg/g hígado) | 3,55 ± 0,57 | 5,11 ± 1,3a (×1,4) |
| Triglicéridos plasmáticos (mg/dl) | 111 ± 28 | 153 ± 19b (×1,37) |
| Leptina (ng/ml) | 1,59 ± 0,45 | 2,97 ± 1,30a (×1,9) |
| Adiponectina (μg/ml) | 3,66 ± 0,88 | 6,36 ± 0,9b (×1,7) |
ABC: área bajo la curva.
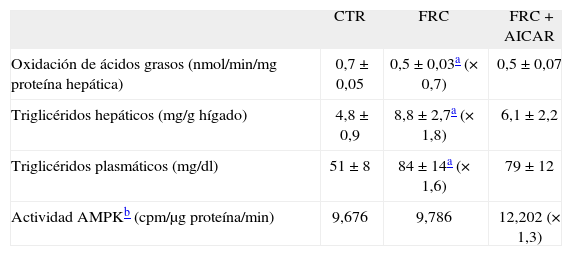
Para determinar el papel de la actividad AMPK en el control de la oxidación de ácidos grasos hepática, administramos AICAR a un grupo de ratas macho suplementadas con fructosa líquida. AICAR es un análogo del AMP capaz de activar de forma directa AMPK18,19. Aunque la administración de AICAR produjo un incremento de un 30% en la actividad hepática de AMPK, no produjo ninguna modificación en la hipertrigliceridemia y el déficit de oxidación de ácidos grasos inducido por fructosa (tabla 2).
Efecto del AICAR en la actividad AMPK, la hipertrigliceridemia y la oxidación hepática de los ácidos grasos
| CTR | FRC | FRC + AICAR | |
| Oxidación de ácidos grasos (nmol/min/mg proteína hepática) | 0,7 ± 0,05 | 0,5 ± 0,03a (×0,7) | 0,5 ± 0,07 |
| Triglicéridos hepáticos (mg/g hígado) | 4,8 ± 0,9 | 8,8 ± 2,7a (×1,8) | 6,1 ± 2,2 |
| Triglicéridos plasmáticos (mg/dl) | 51 ± 8 | 84 ± 14a (×1,6) | 79 ± 12 |
| Actividad AMPKb (cpm/μg proteína/min) | 9,676 | 9,786 | 12,202 (×1,3) |
AICAR: 1 β-D ribofuranósido de 5-aminoimidazol 4-carboxamida; AMPK: AMP activada proteína cinasa; CTR: ratas control; FRC: ratas suplementadas con fructosa en forma líquida.
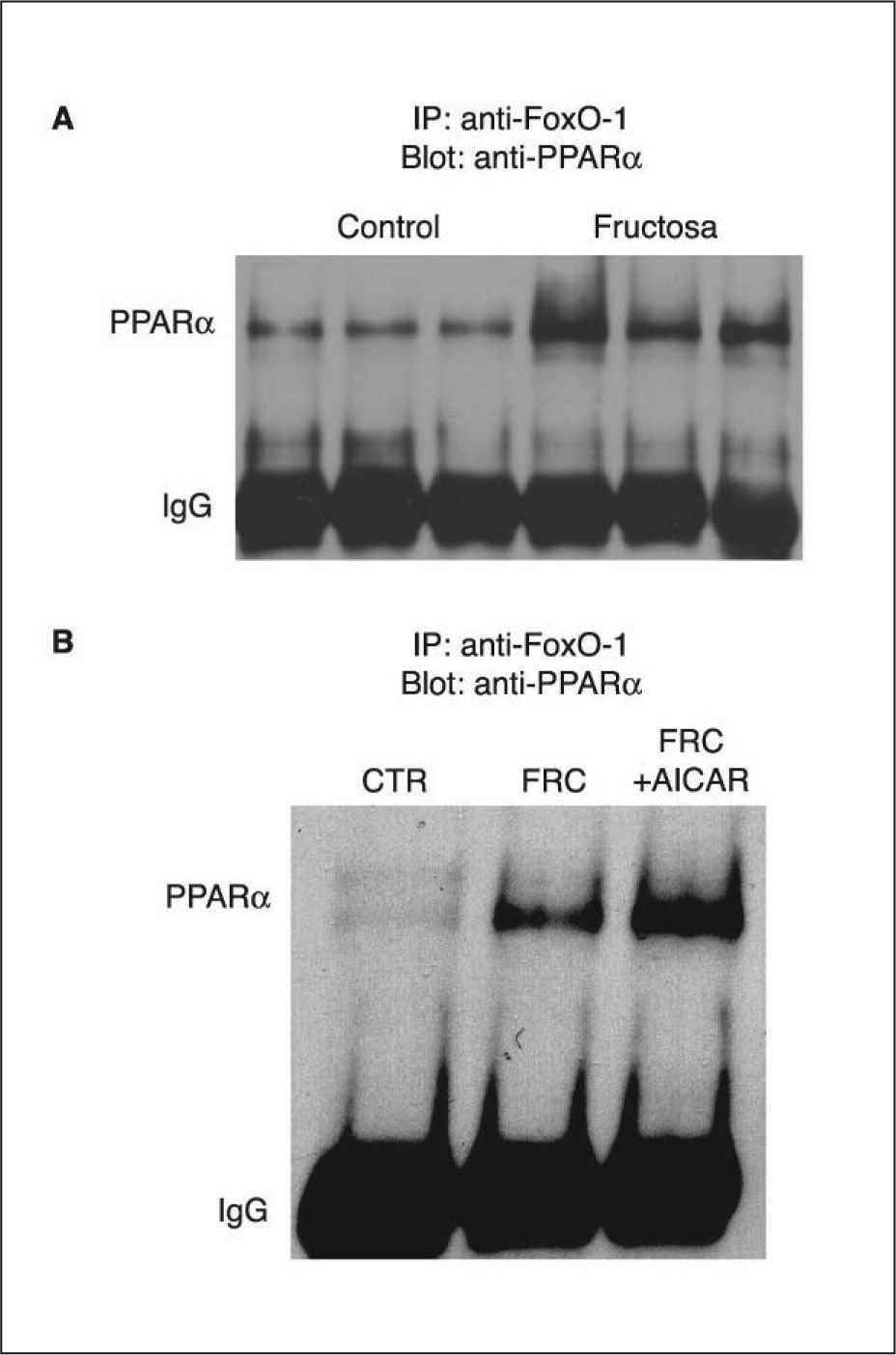
La fructosa reduce la actividad del receptor alfa del activador-proliferador del peroxisoma (PPARα) hepática8. Qu et el20 han demostrado que hámsters alimentados con una dieta sólida con un 60% de fructosa presentan un incremento en la forma activa del factor nuclear FoxO1. La activación de FoxO1 se relacionó con un descenso en el aclaramiento plasmático de triglicéridos en estos animales, y se revirtió con la administración de un agonista PPARα, el fenofibrato. El mecanismo encargado de este efecto fue la actividad de transrepresión de PPARα sobre la actividad transcripcional de FoxO1, tras la interacción física entre las formas activadas de ambos receptores20. Dado que los mecanismos de transrepresión pueden ser bidireccionales16,18, determinamos si se producía una situación similar en el hígado de ratas suplementadas con fructosa en forma líquida al 10% p/v. Como se puede apreciar en la figura 1A, experimentos de coinmunoprecipitación indicaron que la administración de fructosa en forma líquida incrementó la formación de complejos PPARα-FoxO1 en el hígado, en comparación con los animales control. Este efecto de transrepresión por interacción física de FoxO1 sobre PPARα sería la causa directa de la reducción en la actividad hepática de oxidación de ácidos grasos producida por la ingestión de fructosa. FoxO1 es inactivado por fosforilación en un residuo de Ser a través de la actividad proteína cinasa B o Akt21. A su vez, Akt es activada por fosforilación en un residuo de Ser por la cinasa activada de JAK-2. En consecuencia, el exceso de actividad FoxO1 derivaría también del déficit de señalización de la leptina a través de JAK-2 que hemos demostrado previamente (JAK-2). Es interesante señalar que la administración de AICAR no fue capaz de modificar la interacción entre FoxO1 y PPARα (fig. 1B).
 y FoxO1 en muestras hepáticas de ratas suplementadas con fructosa (FRC). A. Extractos nucleares de 3 controles (CTR) y de 3 FRC que se sometieron a inmunoprecipitación con anticuerpo anti-FoxO1 asociado a partículas de proteína A agarosa. Los inmunoprecipitados se sometieron a SDS-PAGE y se inmunorrevelaron con anticuerpo anti-PPARα. Muestras del resto de animales utilizados en el estudio proporcionaron resultados similares. B. Extractos nucleares obtenidos por muestreo de cada muestra individual de ratas CTR, FRC y tratadas con 1 β-D ribofuranósido de 5-aminoimidazol 4-carboxamida (AICAR) (FRC + AICAR) se trataron tal como se describe en el apartado A.")
Coinmunoprecipitación del receptor alfa del activador-proliferador del peroxisoma (PPARα) y FoxO1 en muestras hepáticas de ratas suplementadas con fructosa (FRC). A. Extractos nucleares de 3 controles (CTR) y de 3 FRC que se sometieron a inmunoprecipitación con anticuerpo anti-FoxO1 asociado a partículas de proteína A agarosa. Los inmunoprecipitados se sometieron a SDS-PAGE y se inmunorrevelaron con anticuerpo anti-PPARα. Muestras del resto de animales utilizados en el estudio proporcionaron resultados similares. B. Extractos nucleares obtenidos por muestreo de cada muestra individual de ratas CTR, FRC y tratadas con 1 β-D ribofuranósido de 5-aminoimidazol 4-carboxamida (AICAR) (FRC + AICAR) se trataron tal como se describe en el apartado A.
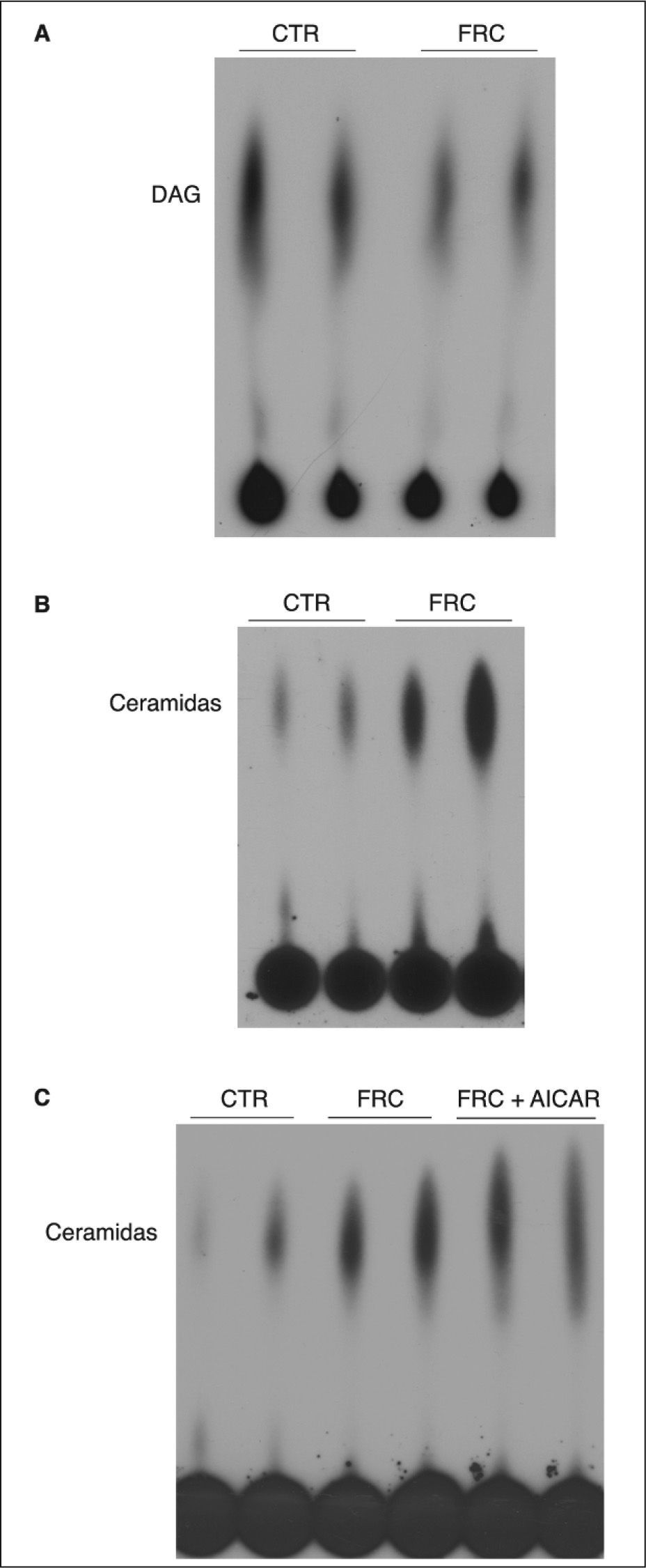
El ácido palmítico es un precursor de la síntesis de novo de ceramidas22. Dado que la fructosa reduce la oxidación hepática de ácidos grasos e incrementa su síntesis, determinamos si el exceso de ácidos grasos hepáticos comportaba un incremento en la síntesis no sólo de triglicéridos, sino también de otras especies lipídicas. Como se observa en la figura 2A, aunque la cantidad hepática de diacilglicerol fue similar entre las ratas control y las tratadas con fructosa, sí que se produjo un incremento marcado en el contenido hepático de ceramida en las ratas suplementadas con fructosa (fig. 2B). Por otra parte, el tratamiento con AICAR no redujo de forma apreciable el contenido hepático de ceramida en los animales suplementados con fructosa (fig. 2C).
 y ceramida en hígados de ratas suplementadas con fructosa (FRC). Se prepararon extractos lipídicos y se valoró la presencia de DAG y ceramida, como se detalla en el apartado Materiales y métodos. Se muestran fosfoimágenes del DAG (A) y la ceramida (B y C) presente en muestras hepáticas representativas, obtenidas por muestreo de 6 muestras individuales de ratas CTR, FRC y tratadas con 1 β-D ribofuranósido de 5-aminoimidazol 4-carboxamida (AICAR) (FCR + AICAR). CTR: control.")
Valores de diacilglicerol (DAG) y ceramida en hígados de ratas suplementadas con fructosa (FRC). Se prepararon extractos lipídicos y se valoró la presencia de DAG y ceramida, como se detalla en el apartado Materiales y métodos. Se muestran fosfoimágenes del DAG (A) y la ceramida (B y C) presente en muestras hepáticas representativas, obtenidas por muestreo de 6 muestras individuales de ratas CTR, FRC y tratadas con 1 β-D ribofuranósido de 5-aminoimidazol 4-carboxamida (AICAR) (FCR + AICAR). CTR: control.
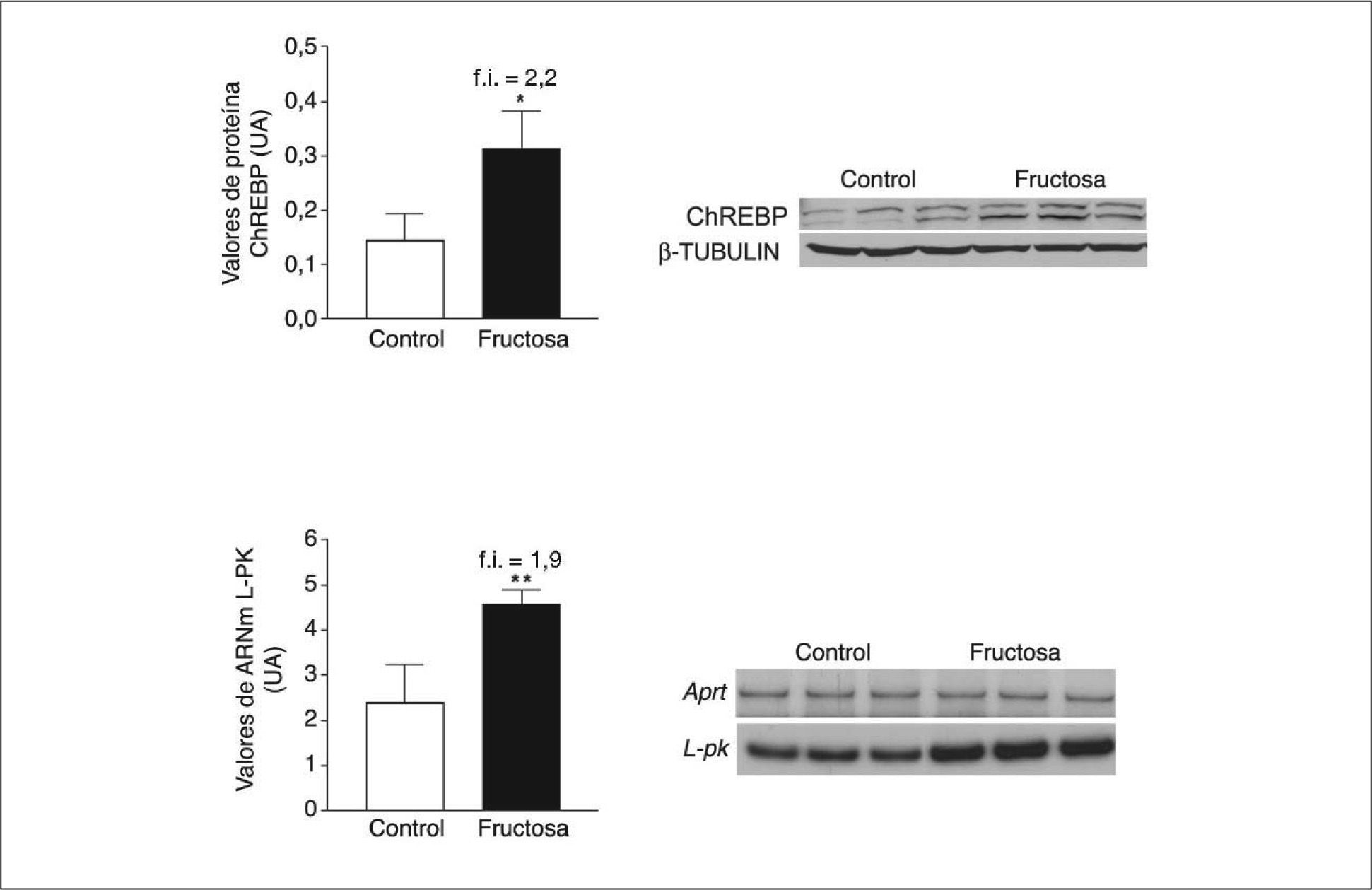
Las ceramidas se comportan como activadores de la PP2A23. La fructosa y otros hidratos de carbono, a través de un metabolito común, la xilulosa-5-fosfato, son también potentes activadores de la PP2A24. Aunque la expresión de la proteína de unión al elemento de respuesta a hidratos de carbono (ChREBP), controlada por PP2A, y de la L-piruvato cinasa24,25, gen diana de ChREBP, se vio incrementada en el hígado de las ratas alimentadas con fructosa (fig. 3), no fuimos capaces de detectar ningún incremento en la expresión de la subunidad catalítica de PP2A.
 en muestras hepáticas de ratas suplementadas con fructosa. A. Gráfica de los valores proteicos de ChREBP en tejido hepático. Cada barra representa la media ± desviación estándar para n = 6 animales por grupo de tratamiento. En la parte derecha de la gráfica se puede observar la autorradiografía representativa del ensayo de Western-blot, en el que se observan las bandas correspondientes a 3 ratas control y 3 ratas suplementadas con fructosa. La cantidad de proteína cargada se confirmó por el método de Bradford, y la uniformidad de carga en cada línea mediante la determinación de la señal de la proteína β-tubulina. B. Valores relativos de ácido ribonucleico mensajero (ARNm) para la L-PK en muestras hepáticas control y fructosa. Cada valor representa la media ± desviación estándar de n = 6 animales por grupo, expresado en unidades arbitrarias (UA). *p < 0,05. **p < 0,01.")
Expresión de ChREBP y L-piruvato cinasa (L-PK) en muestras hepáticas de ratas suplementadas con fructosa. A. Gráfica de los valores proteicos de ChREBP en tejido hepático. Cada barra representa la media ± desviación estándar para n = 6 animales por grupo de tratamiento. En la parte derecha de la gráfica se puede observar la autorradiografía representativa del ensayo de Western-blot, en el que se observan las bandas correspondientes a 3 ratas control y 3 ratas suplementadas con fructosa. La cantidad de proteína cargada se confirmó por el método de Bradford, y la uniformidad de carga en cada línea mediante la determinación de la señal de la proteína β-tubulina. B. Valores relativos de ácido ribonucleico mensajero (ARNm) para la L-PK en muestras hepáticas control y fructosa. Cada valor representa la media ± desviación estándar de n = 6 animales por grupo, expresado en unidades arbitrarias (UA). *p < 0,05. **p < 0,01.
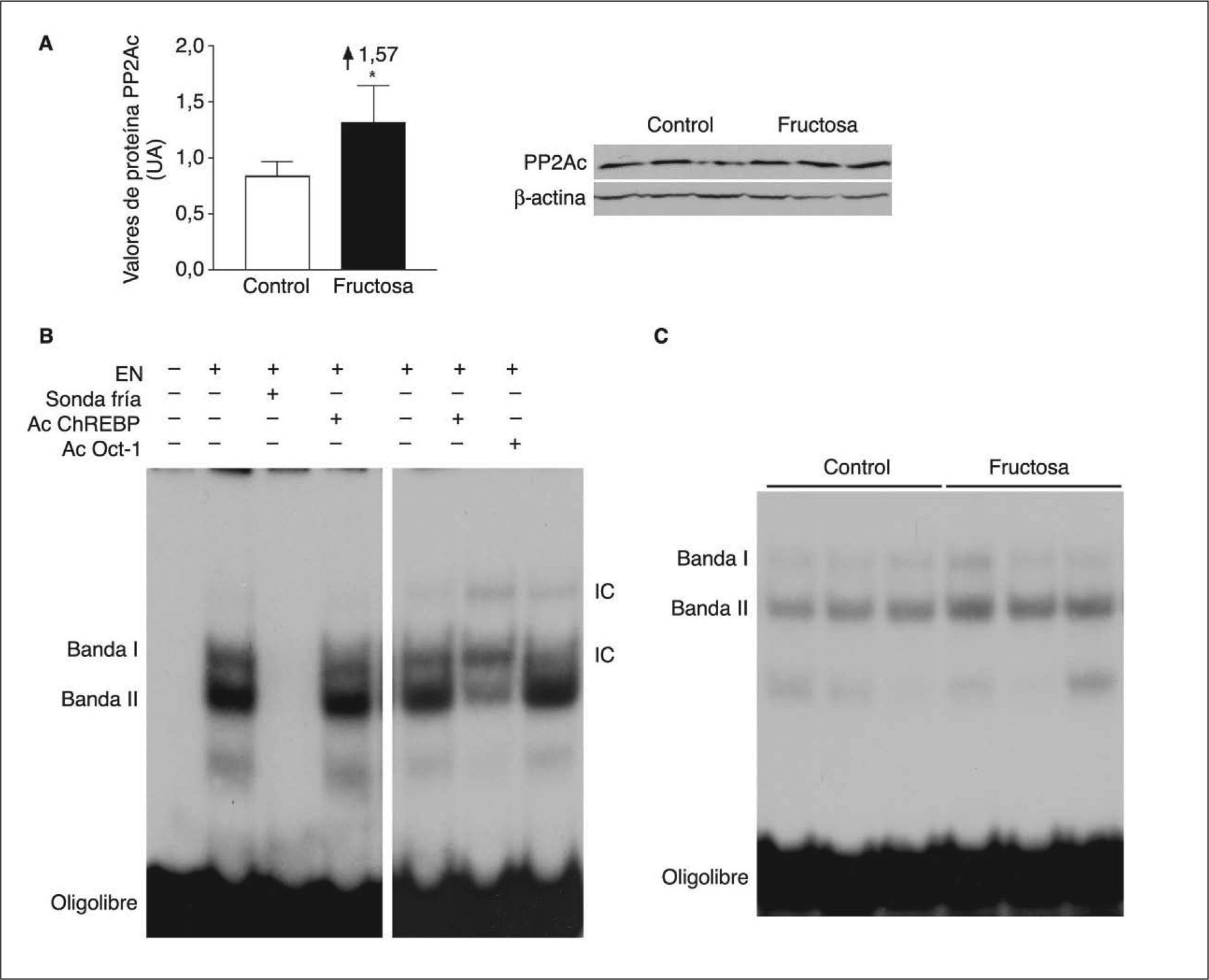
Para evitar el posible efecto en los valores hepáticos de xilulosa-5-fosfato de las 2h de ayuno, determinamos la expresión de la subunidad catalítica de PP2A y la actividad de unión del ChREBP en muestras hepáticas de ratas suplementadas con fructosa y sacrificadas sin ayuno previo. En estas condiciones, la administración de fructosa produjo un incremento en la expresión de la subunidad catalítica de PP2A (×1,6) (fig. 4A). La incubación de extractos nucleares hepáticos con un oligonuclótido que mimetiza el elemento de respuesta a ChREBP produjo 2 bandas específicas, cuya intensidad disminuyó en presencia de un exceso de oligonucleótido no marcado (fig. 4B). Asimismo, ambas bandas contenían proteína ChREBP, ya que se vieron superretardadas cuando los extractos se incubaron en presencia de un anticuerpo específico anti-ChREBP. Como se puede apreciar en la figura 4B, la intensidad de la banda II se incrementó de una forma marcada (×1,4) en los extractos nucleares provenientes de ratas suplementadas con fructosa. En conjunto, estos resultados indican que la ingestión de fructosa en forma líquida incrementa la actividad PP2A hepática.
 y 3 animales suplementados con fructosa (barra negra). La cantidad de proteína cargada se confirmó por el método de Bradford, y la uniformidad de carga en cada línea mediante la determinación de la señal de la proteína β-actina. *p < 0,05. B. Ensayo de retardación electroforética (EMSA) en el que se muestra la unión de extractos nucleares hepáticos (EN) a un oligonucleótido que mimetiza en el elemento de respuesta a ChREBP y la formación de 2 bandas específicas que desaparecen en presencia de un exceso de sonda fría. Las bandas I y II contienen proteína ChREBP al ser superretardadas en presencia de un anticuerpo específico anti-ChREBP. El anticuerpo anti-Oct-1 se usó para demostrar que los cambios en la movilidad de las bandas no se debieron a una interferencia inespecífica por la presencia de inmunoglobulinas en el medio de incubación. C. EMSA en el que se muestra la unión a un oligonucleótido ChREBP de extractos nucleares hepáticos, obtenidos de ratas control y suplementadas con fructosa. IC: inmunocomplejo.")
Expresión de la subunidad catalítica de PP2A y actividad de unión de ChREBP en ratas suplementadas con fructosa y sacrificadas sin ayuno previo. Western-blot de la subunidad catalítica, activa, de PP2A en muestras hepáticas de ratas control y suplementadas con fructosa. A. En la parte izquierda de la figura, un diagrama de barras muestra los valores de proteína PP2A, expresados como la media ± desviación estándar de los valores obtenidos en muestras de 3 controles (barra blanca) y 3 animales suplementados con fructosa (barra negra). La cantidad de proteína cargada se confirmó por el método de Bradford, y la uniformidad de carga en cada línea mediante la determinación de la señal de la proteína β-actina. *p < 0,05. B. Ensayo de retardación electroforética (EMSA) en el que se muestra la unión de extractos nucleares hepáticos (EN) a un oligonucleótido que mimetiza en el elemento de respuesta a ChREBP y la formación de 2 bandas específicas que desaparecen en presencia de un exceso de sonda fría. Las bandas I y II contienen proteína ChREBP al ser superretardadas en presencia de un anticuerpo específico anti-ChREBP. El anticuerpo anti-Oct-1 se usó para demostrar que los cambios en la movilidad de las bandas no se debieron a una interferencia inespecífica por la presencia de inmunoglobulinas en el medio de incubación. C. EMSA en el que se muestra la unión a un oligonucleótido ChREBP de extractos nucleares hepáticos, obtenidos de ratas control y suplementadas con fructosa. IC: inmunocomplejo.
Recientemente, nuestro grupo ha demostrado que la administración de fructosa al 10% en el agua de bebida a ratas induce hipertrigliceridemia y esteatosis hepática, junto con un estado de resistencia hepática a la leptina y de hiperleptinemia8,10, y que este estado de resistencia es la causa de la falta de actividad de AMPK y PPARα. Además, hemos proporcionado pruebas de que la diferencia importante entre la ingestión de cantidades similares de fructosa y glucosa en el agua de bebida es el hecho que, mientras ambos azúcares incrementan de forma similar la lipogenia hepática, sólo el primero es capaz de reducir de forma significativa la actividad hepática de β-oxidación de los ácidos grasos. En el presente trabajo, proporcionamos datos experimentales que revelan un posible mecanismo capaz de explicar las alteraciones metabólicas que aparecen en el ámbito hepático cuando se suplementa la dieta de ratas con fructosa al 10% en el agua de bebida.
La reducción en la oxidación hepática de ácidos grasos puede atribuirse a la falta de activación por leptina de 2 proteínas implicadas en el control del catabolismo de los ácidos grasos: la enzima AMPK y el receptor nuclear PPARα26,27. Los resultados obtenidos tras el tratamiento con AICAR indican que, al menos en el tejido hepático, el control de la oxidación de ácidos grasos lo ejerce de forma predominante PPARα, ya que la activación de AMPK en el hígado de los animales tratados con AICAR no revirtió el déficit oxidativo, ni la hipertrigliceridemia.
En cualquier caso, la falta de activación de AMPK y PPARα en el hígado de las ratas suplementadas con fructosa se relaciona con un déficit generalizado en la fosforilación de residuos de Ser/Thr en proteínas clave en la transducción de la señal de la leptina28. Entre ellas, cabe señalar la falta de activación de JAK-2, que redunda en un déficit de activación de Akt y, por consiguiente, en un estado de hiperactivación del factor de transcripción FoxO128–30. Esta situación es finalmente la causa de la transrepresión de la actividad de PPARa, debido a su interacción física con la forma no fosforilada, activa de FoxO1.
El conocimiento existente sobre el metabolismo de hidratos de carbono indicaría que la fosfatasa de residuos de Ser/Thr PP2A es la causa probable de esta situación. El metabolismo de la fructosa produce xilulosa-5-fosfato, un activador de PP2A25. Aunque no detectamos incrementos en la forma activa de PP2A en el hígado de las ratas suplementadas con fructosa, este hecho se debió probablemente a las 2h de ayuno a las que se sometieron los animales. Cuando se valoró la expresión de la forma activa de PP2A y la actividad de unión del ChREBP, cuya activación depende directamente de PP2A24,25 en muestras hepáticas de ratas suplementadas con fructosa y no sometidas a ayuno, los resultados obtenidos indican de hecho una activación de PP2A por la ingestión de fructosa en forma líquida. Igualmente, el hecho de que los hígados de estos animales se encuentren enriquecidos en ceramida, otra molécula activadora de PP2A23, concuerda con la presencia de PP2A activada. Finalmente, la presencia de ácidos grasos saturados, que presumiblemente se acumularían en el tejido hepático de los animales suplementados con fructosa, debido a la inhibición de la oxidación y el incremento de la síntesis de estos lípidos, contribuiría también a la activación de PP2A31. Se ha descrito que la actividad PP2A reprime la expresión de PPARα y la actividad de Akt y AMPK31–33, efectos que contribuirían a las alteraciones metabólicas inducidas por la fructosa. El hecho de que la fructosa se metabolice de forma mayoritaria en el hígado –mientras que la glucosa se distribuye y metaboliza en otros tejidos– explicaría por qué, a pesar de que la glucosa también origine xilulosa-5-fosfato25, no sea capaz de inducir hiperpleptinemia y resistencia hepática a la leptina cuando se ingiere en cantidades equivalentes con la fructosa.
Al CIBERDEM, una iniciativa del Instituto de Salud Carlos III.
Este trabajo ha sido financiado por una beca FEA/SEA Manuel de Oya, investigación en Nutrición, convocatoria 2007 y por los proyectos FIS PI070875//PI060247.