





La resistencia a la insulina se relaciona con la presencia de un proceso inflamatorio de baja intensidad, caracterizado por la activación crónica del factor nuclear kappa B (NF-κB) y la producción de citocinas proinflamatorias en tejido adiposo blanco. Recientemente, se ha propuesto que el receptor activado por proliferadores peroxisómicos β/δ (PPARβ/δ) puede llegar a convertirse en una diana para el tratamiento de la resistencia a la insulina. Sin embargo, se desconoce si la activación de este receptor nuclear previene el proceso inflamatorio en adipocitos.
Métodos y resultadosLa activación de PPARβ/δ por un ligando específico, GW501516, evitó el aumento en la expresión y la secreción de interleucina (IL) 6 inducida por lipopolisacárido (LPS) en adipocitos 3T3-L1. Este efecto se asoció con la capacidad del agonista de PPARβ/d para inhibir la activación del NF-κB inducida por LPS. Por otro lado, la expresión de IL-6 y la actividad del NF-κB estaban incrementadas en el tejido adiposo de ratones PPARβ/δ−/−, en comparación con ratones wild-type, hecho que indica que este receptor regula la activación de NF-κB y de sus genes diana. Puesto que se ha relacionado la activación de la vía ERK1/2 con la activación de NF-κB, determinamos el efecto de PPARβ/d en la activación de esta vía. El tratamiento con GW501516 evitó la fosforilación de ERK1/2 inducida por LPS, mientras que el tejido adiposo blanco de ratones PPARβ/δ−/− mostró un incremento considerable en los valores de fosforilación de esta cinasa.
ConclusionesEstos resultados parecen indicar que la activación de PPARβ/δ bloquea el incremento de la producción de citocinas inflamatorias en adipocitos mediante la prevención de la activación de NF-κB por ERK1/2. Esta acción de PPARβ/δ puede contribuir a prevenir la resistencia a la insulina.
Insulin resistance is associated with a low-grade systemic inflammatory response characterized by NF-κB activation and pro-inflammatory cytokine production from white adipose tissue. Recently PPARβ/δ has been proposed as a potential treatment for insulin resistance. However, it is presently unknown whether PPARβ/δ activation prevents this process in adipocytes.
Methods and resultsPPARβ/δ activation by GW501516 blocked LPS-induced IL-6 expression and secretion by 3T3-L1 adipocytes. This effect was associated with the capacity of GW501516 to prevent LPS-induced NF-κB activation. In addition, IL-6 expression and NF-κB DNA-binding activity was higher in white adipose tissue from PPARβ/δ-null mice than in wild-type mice, fact that suggests that this receptor regulates NF-κB activation and its target genes. Since the ERK1/2 pathway has been involved in NF-κB activation, we explored the effect of PPARβ/δ activation on this pathway. GW501516 treatment prevented ERK1/2-phosphorylation by LPS, whereas white adipose tissue from PPARβ/δ-null mice showed increased phospho-ERK1/2 levels.
ConclusionsThese findings indicate that PPARβ/δ activation blocks enhanced cytokine production in adipocytes by preventing NF-κB activation via ERK1/2. This PPARβ/δ effect may contribute to prevent insulin resistance.
La obesidad visceral se relaciona con la presencia de un proceso inflamatorio crónico de intensidad baja, que puede estar implicado en el desarrollo de la resistencia a la insulina y enfermedades cardiovasculares1. Además, distintos modelos de obesidad presentan un aumento en la expresión de distintas citocinas proinflamatorias (como el factor de necrosis tumoral alfa [TNF-α], la interleucina [IL] 1, proteína quimiotáctica de monocitos [MCP] 1 e IL-6) en el adipocito2–4. De estos mediadores inflamatorios, la IL-6 es la que presenta una correlación mayor con el desarrollo de la resistencia a la insulina y la diabetes mellitus tipo 25–7. La expresión de estos marcadores de inflamación por parte del adipocito durante procesos de obesidad y de resistencia a la insulina se ha relacionado con la activación del factor de transcripción proinflamatorio factor nuclear kappa B (NF-κB)4. Este factor de transcripción puede activarse mediante una serie de estímulos exógenos, como el lipopolisacárido (LPS), de las bacterias gramnegativas y endógenos, como los ácidos grasos saturados. La activación de NF-κB por estos componentes es mediada a través de la activación del receptor TLR4 (Toll-Like Teceptor-4)8. Cuando este receptor se activa, selecciona el factor de diferenciación mieloide 88, (MyD88, Myeloid Differentiation Factor-88), que a su vez activa al receptor de IL-1 asociado a cinasa (IRAK, del inglés interleukin-1 receptor-associated kinase), y desencadena una serie de procesos que conducen a la activación de las IκB cinasas (IKK) y, por tanto, a la activación de NF-κB y al desarrollo de la respuesta inflamatoria.
En estudios recientes se indica que el proceso inflamatorio, inducido por obesidad o por el consumo de dietas con contenido alto en lípidos, causa resistencia a la insulina a través de mecanismos que implican la activación de TLR49. La activación de TLR4 es fundamental para el desarrollo de la respuesta inflamatoria inducida por ácidos grasos saturados en adipocitos y macrófagos, ya que esta respuesta no se desarrolla en ausencia de este receptor9. De este modo el incremento en la lipólisis observado en el tejido adiposo durante estados de resistencia a la insulina, se relaciona con la producción de ácidos grasos saturados y la activación de TLR4, lo cual desencadena el proceso inflamatorio10. Por otro lado, las dietas con un contenido alto en grasas incrementan el LPS plasmático y de este modo potencian el desarrollo del proceso inflamatorio11. Además, ratones deficientes en el receptor para el LPS (mutantes CD14) mostraban hipersensibilidad a la insulina y cuando se sometían a una dieta rica en grasas aparecía retrasado el desarrollo de resistencia a la insulina, obesidad y diabetes mellitus11.
En los últimos años, el receptor activado por proliferadores peroxisómicos de tipo β/δ (PPARβ/δ) se ha propuesto como una diana potencial para el tratamiento de la resistencia a la insulina12. Los receptores activados por proliferadores (PPAR) son factores de transcripción activados por ligando que pertenecen a la superfamilia de receptores nucleares. En su forma activa dimerizan con receptores retinoides X (RXR, del inglés retinoid X receptor) e interaccionan con secuencias consenso situadas en la región promotora de los genes que regulan, formadas por repeticiones directas de secuencias hexaméricas separadas por un nucleótido13. Una vez el heterodímero PPAR/RXR es activado por ligando, se produce un cambio conformacional que permite el intercambio de proteínas correpresoras y coactivadoras, encargadas de seleccionar toda la maquinaria necesaria para el inicio de la transcripción. Además, los PPAR pueden suprimir la inflamación a través de distintos procesos, como reducir la liberación de marcadores de inflamación, o estabilizar complejos represores en los promotores de genes inflamatorios14–17. De los 3 miembros de esta familia encontrados en mamíferos, PPARα y PPARγ actúan como dianas para fármacos hipolipemiantes (fibratos) y antidiabéticos (tiazolidindionas), respectivamente. La activación del PPARβ/δ por ligandos de elevada afinidad incrementa el catabolismo de los ácidos grasos en adipocitos y músculo esquelético12. Sin embargo, se desconoce si PPARβ/δ previene el proceso inflamatorio en adipocitos. Por ello, en este trabajo examinamos si la activación de PPARβ/δ por GW501516 prevenía el proceso inflamatorio inducido por LPS en adipocitos. Los resultados obtenidos indican que el agonista de PPARβ/δ previene la inducción en la expresión y secreción de IL-6 en adipocitos, hecho relacionado con la capacidad de PPARβ/δ de prevenir la activación de NF-κB por LPS en estas células. Además, la expresión de IL-6 y la actividad de unión a ADN de NF-κB se encontraban incrementadas en el tejido adiposo de ratones deficientes para el gen PPARβ/δ (PPARβ/δ−/−) comparado con el tejido adiposo de ratones wild-type. Los datos de este estudio también indican que PPARβ/δ puede prevenir el proceso inflamatorio en adipocitos mediado por NF-κB gracias a su capacidad para inhibir la fosforilación de ERK1/2. En resumen, los resultados de este trabajo indican que la activación de PPARβ/δ puede ser útil para prevenir el proceso inflamatorio en adipocitos y, en consecuencia, la resistencia a la insulina asociada a este proceso.
MétodosReactivosEl ligando de PPARβ/δ, GW501516, se obtuvo de Biomol Research Labs (Plymouth Meeting, PA [Estados Unidos]). El resto de reactivos proceden de Sigma (St. Louis, MO [Estados Unidos]).
Cultivo celularLa línea de adipocitos 3T3-L1 (American Type Culture Collection) se mantuvo hasta confluencia en DMEM (del inglés Dulbecco's modified Eagle's medium) suplementado con 10% de suero bovino fetal, 500μmol/l metilisobutilxantina, 0,25μmol/ l dexametasona y 10μg/ml de insulina durante 48h. Las células se incubaron en DMEM con un 10% de FBS con insulina durante 8 días. Los adipocitos se incubaron entonces durante 96h con 0,5μmol/l de GW501516 en presencia y ausencia de 100ng/ml de LPS, durante una o 24h. Después de la incubación, el ácido ribonucleico (ARN), la proteína total y los extractos de proteína nuclear se extrajeron de los adipocitos, como se explicará posteriormente. Los inhibidores fueron adicionados a las células 30min antes de la estimulación con LPS. La secreción de IL-6 fue evaluada mediante la técnica de ELISA (Amersham Biosciences, Little Chalfont [Reino Unido]) en el medio de cultivo celular.
AnimalesEl tejido adiposo procedente de ratones deficientes en el gen PPARβ/δ (PPARβ/δ-/-) fue cedido por el Dr. Walter Wahli, el cual previamente generó este modelo a partir de ratones C57BL/618. Como control se utilizaron ratones wild type con el mismo background genético.
Preparación y análisis del ácido ribonucleicoLos valores de ARN mensajero (ARNm) se determinaron por transcriptasa reversa-reacción en cadena de la polimerasa (RTPCR), tal como se ha descrito previamente19. El ARN total fue aislado utilizando el reactivo Ultraspec (Biotecx, Houston, TX [Estados Unidos]), siguiendo las indicaciones del fabricante. Las secuencias utilizadas para la amplificación se muestran en la tabla 1. Previamente se determinaron las condiciones óptimas de trabajo para evitar trabajar en estado de saturación. La intensidad de las bandas se cuantificó por densitometría (Vilbert Lourmat Imaging). Los resultados de la expresión relativa de los distintos ARNm se normalizaron utilizando el gen Aprt como control interno.
Primers utilizados para la reacción en cadena de la polimerasa (PCR)
| Gen | Secuencia de los primers | Producto de PCR (pb) | Ciclos de amplificación |
| APRT | Forward: 5'-GCCTCTTGGCCAGTCACCTGA-3'Reverse: 5'-CCAGGCTCACACACTCCACCA-3' | 329 pb | 23 |
| IL-6 | Forward: 5'-TCCAGCCAGTTGCCTTCTTGG-3'Reverse: 5'-TCTGACAGTGCATCATCGCTG-3' | 229 pb | 35 |
| MCP-1 | Forward: 5'-GGGCCTGTTGTTCACAGTTGC-3'Reverse: 5'-GGGACACCTGCTGCTGGTGAT-3' | 157 pb | 23 |
Los extractos nucleares se extrajeron tal como se ha descrito previamente19. Los ensayos de retardación de la movilidad electroforética (EMSA) se realizaron utilizando oligonucleótidos de doble cadena (Promega, Madison, WI [Estados Unidos]) para las secuencias consenso de unión de NF-κB (5'-AGTTGAGGGGACTTTCCCAGGC-3'). Los oligonucleótidos se marcaron con [γ-32P]ATP (3,000Ci/mmol a 10mCi/ml) y purificados en columnas Nick (Amersham), para eliminar el ATP no unido a las sondas. Se incubaron 8 microgramos de extractos de proteína nuclear durante 10min en hielo en tampón de unión (10mmol/l de Tris–HCl, pH 8,0, 25mmol/l de cloruro potásico [KCl], 0,5mmol/l de DTT, 0.1mmol/l de ácido etilendiaminotetraacético [EDTA], pH 8,0, 5% de glicerol, 5mg/ml de BSA, y 50μg/ml de Poly[dI-dC]), en un volumen final de 15μl. La sonda marcada (60.000cpm) se añadió y la reacción se incubó durante 15min a 4 °C. Se realizaron ensayos de competición con un exceso de sonda fría y de superretardación con un anticuerpo específico contra p65. Los complejos proteína-ADN se separaron a 4 °C en gel de acrilamida al 5% y se sometieron a autorradiografía.
Análisis de Western-blotPara obtener proteína total, las muestras se homogeneizaron en tampón de lisis frío (5mmol/l Tris–HCl, pH 7,4, 1mmol/ l de EDTA, 0,1mmol/l de ortovanadato sódico y 5,4μg/ml de aprotinina). Los homogenados se centrifugaron a 16,700g durante 30min a 4 °C y la concentración de proteína se determinó por el método de Bradford.
Las proteínas (30μg) se separaron en SDS-PAGE al 10% de acrilamida y se transfirieron a membranas de polivilideno (Millipore, Bedford, MA [Estados Unidos]). La immunodetección se realizó utilizando anticuerpos contra la proteína ERK1/2 total y fosforilada (Cell Signaling). La detección se llevó a cabo utilizando el kit de quimioluminiscencia EZ-ECL (Amersham). El tamaño de las proteínas se determinó utilizando un estándar de pesos moleculares (Invitrogen, Barcelona [España]).
Análisis estadísticoLos resultados se expresan como la media ± desviación estándar de 5 experimentos diferentes. La significación estadística se evaluó mediante ANOVA o t de Student, en función del número de grupos analizado, utilizando el programa informático GraphPad Instat, v2.03 (GraphPad Software, San Diego, CA [Estados Unidos]). En los casos que se observaron diferencias significativas, se aplicó el test de comparaciones múltiples Tukey-Kramer. Las diferencias se consideraron como significativas a partir de p < 0,05.
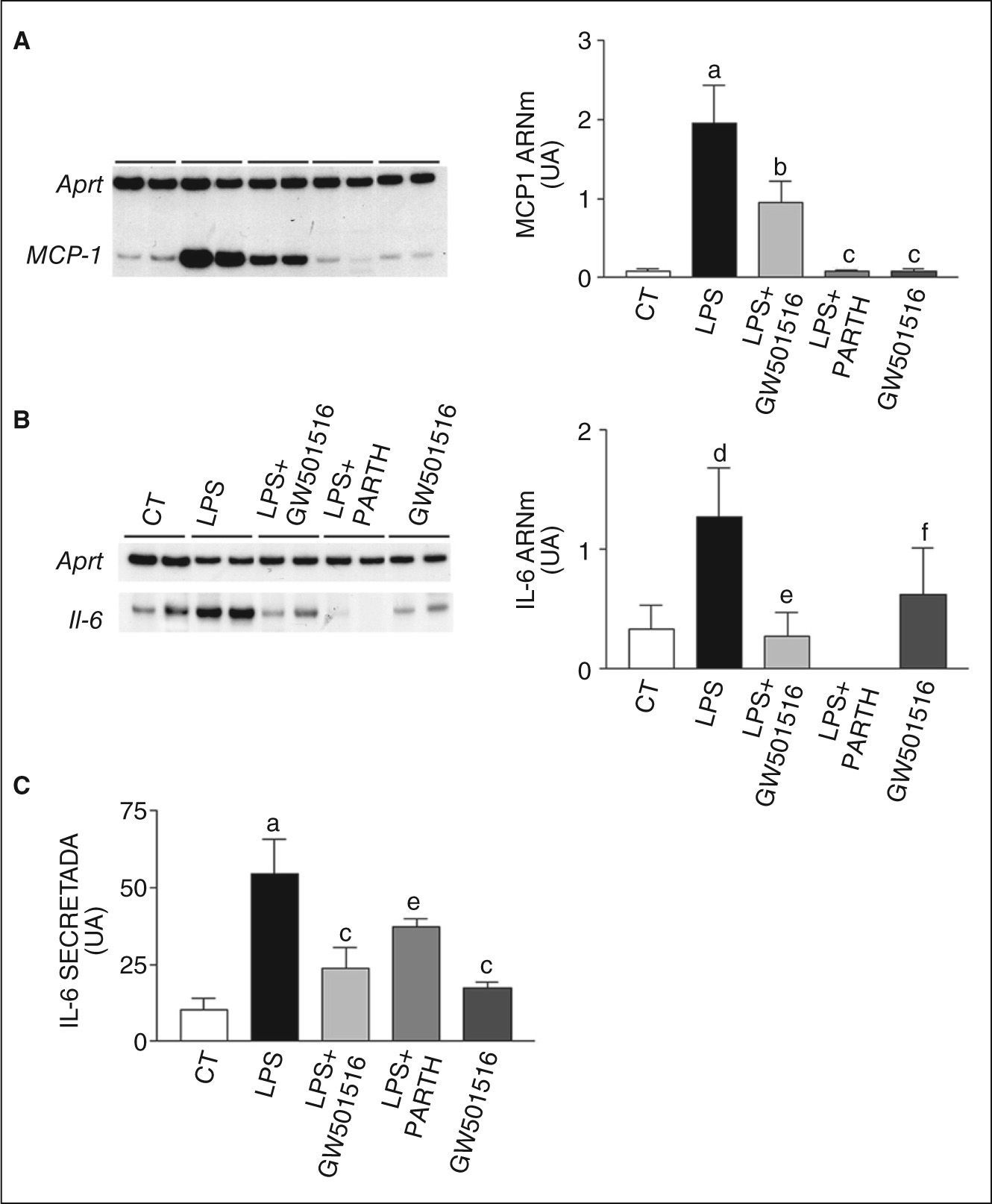
ResultadosLa activación de PPARβ/δ previene el incremento en la expresión y la secreción de IL-6 inducido por LPSAdipocitos 3T3-L1 diferenciados se expusieron durante 96h a 0,5μmol/l de GW501516, un ligando específico para PPARβ/δ, con un valor aparente de Kd de 1nmol/l. Este ligando presenta 1,000 veces más afinidad por PPARβ/δ que por PPAR·y PPARγ20. Después de las 96h de incubación con GW501516, las células se expusieron a 100ng/ml de LPS durante 24h para inducir inflamación. El LPS activa NF-κB a través de los TLR en adipocitos11 e induce resistencia a la insulina11,21. Al evaluar los valores de ARNm de MCP-1 e IL-6, 2 genes regulados por el factor de transcripción proinflamatorio NF-κB, observamos que el tratamiento con LPS incrementaba fuertemente la expresión del primero (inducción de 22 veces; p < 0,001), mientras que las células coincubadas con GW501516 mostraron una reducción significativa de este incremento (reducción del 51%, p < 0,01, frente a células expuestas a LPS) (fig. 1A). Además, en las células coincubadas con 10μmol/l de partenolida, inhibidor específico de NF-κB22, el efecto del LPS se suprimió. El tratamiento con LPS también incrementó los valores de ARNm de IL-6 (inducción de 4,2 veces; p < 0,05), incremento que se bloqueó en presencia de GW501516 y de partenolida (fig. 1B). Asimismo, la estimulación con LPS incrementó 4,2 veces los valores de citocina IL-6 secretada al medio de cultivo (control 10,5 ± 3,7 frente a LPS 55 ± 11ng/ml) (fig. 1C), mientras que en presencia de GW501516 (reducción del 56%; p < 0,001) y partenolida (reducción del 32%; p < 0,05) se observó una reducción significativa de esta secreción.
 GW501516 previene la inducción en la expresión y la secreción de citocinas proinflamatorias inducida por lipopolisacárido (LPS) en adipocitos 3T3-L1. Efecto de GW501516 y parthenolida (PARTH) en la expresión de proteína quimiotáctica de monocitos 1 (MCP-1) (A) e IL-6 (B). La cuantificación de la autorradiografía representativa se encuentra normalizada para los valores de Aprt. C. El efecto de GW501516 sobre la secreción de IL-6 en medio de cultivo fue determinado por ELISA. Los datos se expresan como la media ± desviación estándar de 5 experimentos independientes. ap < 0,001 frente a control. bp < 0,01. cp < 0,001 frente a células expuestas a LPS. dp < 0,05. ep < 0,05. UA: unidades arbitrarias.")
El agonista del receptor activado por proliferadores peroxisómicos β/δ (PPARβ/δ) GW501516 previene la inducción en la expresión y la secreción de citocinas proinflamatorias inducida por lipopolisacárido (LPS) en adipocitos 3T3-L1. Efecto de GW501516 y parthenolida (PARTH) en la expresión de proteína quimiotáctica de monocitos 1 (MCP-1) (A) e IL-6 (B). La cuantificación de la autorradiografía representativa se encuentra normalizada para los valores de Aprt. C. El efecto de GW501516 sobre la secreción de IL-6 en medio de cultivo fue determinado por ELISA. Los datos se expresan como la media ± desviación estándar de 5 experimentos independientes. ap < 0,001 frente a control. bp < 0,01. cp < 0,001 frente a células expuestas a LPS. dp < 0,05. ep < 0,05. UA: unidades arbitrarias.
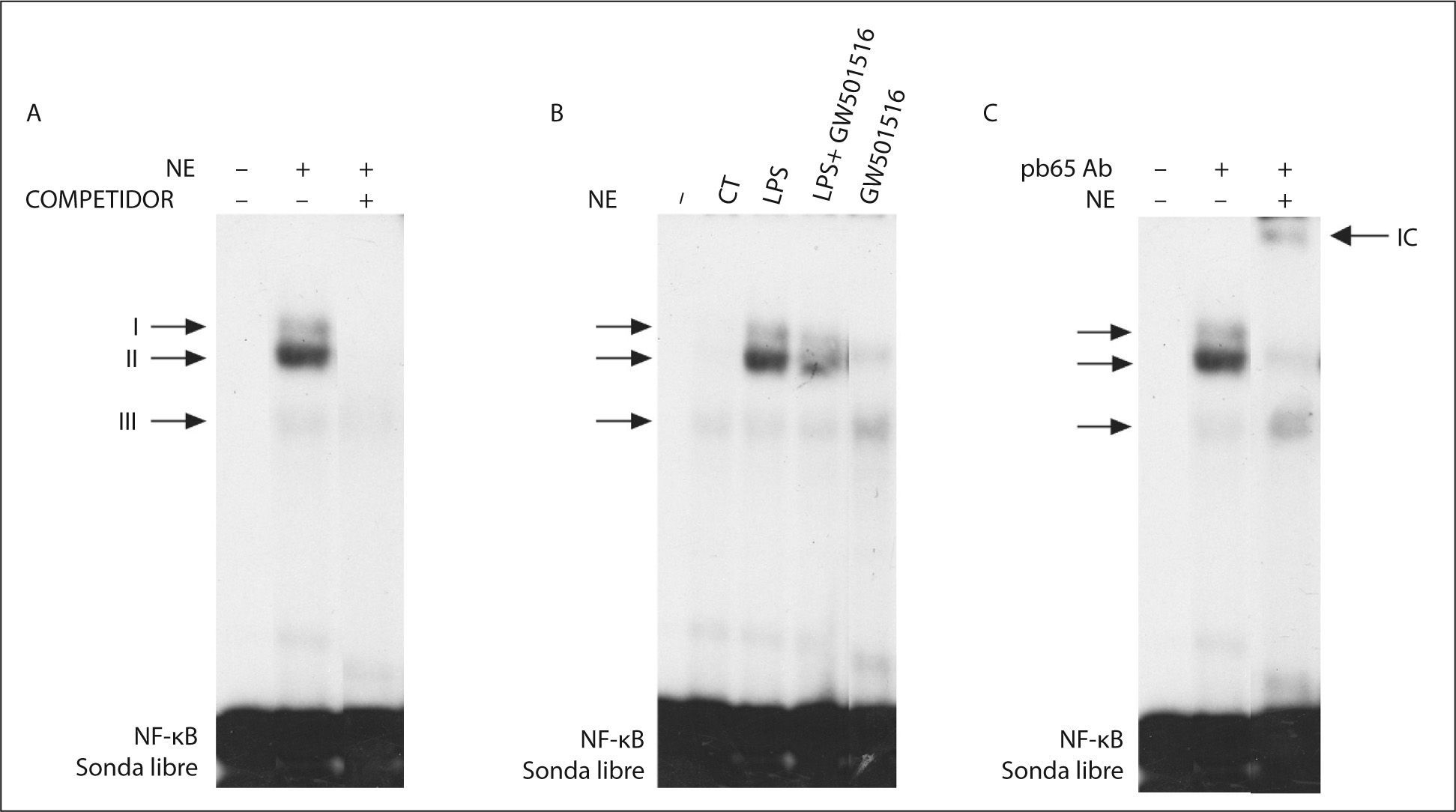
Tal como indican los resultados obtenidos con partenolida, la activación de NF-κB desempeña una papel fundamental en la producción de IL-6 en adipocitos. Para evaluar si GW501516 prevenía la inducción en la expresión de IL-6 provocada por LPS a través de la reducción de la actividad NF-κB, realizamos ensayos de EMSA. El NF-κB formó 3 complejos específicos con proteínas nucleares (complejos I-III), ya que desaparecían al competir con un exceso de sonda fría (fig. 2A). La actividad de unión al ADN de NF-κB se encontraba incrementada en células estimuladas con LPS, mientras que en las células que recibieron el agonista de PPARβ/δ esta actividad estaba reducida (fig. 2B). La adición de un anticuerpo contra la subunidad p65 de NF-κB causó una super-retardación del complejo II, indicando que esta banda contenía principalmente esta subunidad (fig. 2C).
 GW501516 previene la activación de factor nuclear kappa B (NF-κB) inducida por lipopolisacárido (LPS) en adipocitos 3T3-L1. Autorradiografía representativa del ensayo de retardación de la movilidad electroforética realizado con una sonda de NF-κB marcada con 32P y extractos de proteína nuclear. A. Ensayo de competición realizado con un exceso de sonda fría donde se puede apreciar la formación de 3 complejos específicos (I, II y III). B. Efecto del GW501516 en la interacción de la sonda NF-κB con el ácido desoxirribonucleico. C. Ensayo de super-retardación realizado con un anticuerpo específico contra la subunidad p65 de NF-κB.")
El agonista del receptor beta/delta del activador-proliferador del peroxisoma (PPARβ/δ) GW501516 previene la activación de factor nuclear kappa B (NF-κB) inducida por lipopolisacárido (LPS) en adipocitos 3T3-L1. Autorradiografía representativa del ensayo de retardación de la movilidad electroforética realizado con una sonda de NF-κB marcada con 32P y extractos de proteína nuclear. A. Ensayo de competición realizado con un exceso de sonda fría donde se puede apreciar la formación de 3 complejos específicos (I, II y III). B. Efecto del GW501516 en la interacción de la sonda NF-κB con el ácido desoxirribonucleico. C. Ensayo de super-retardación realizado con un anticuerpo específico contra la subunidad p65 de NF-κB.
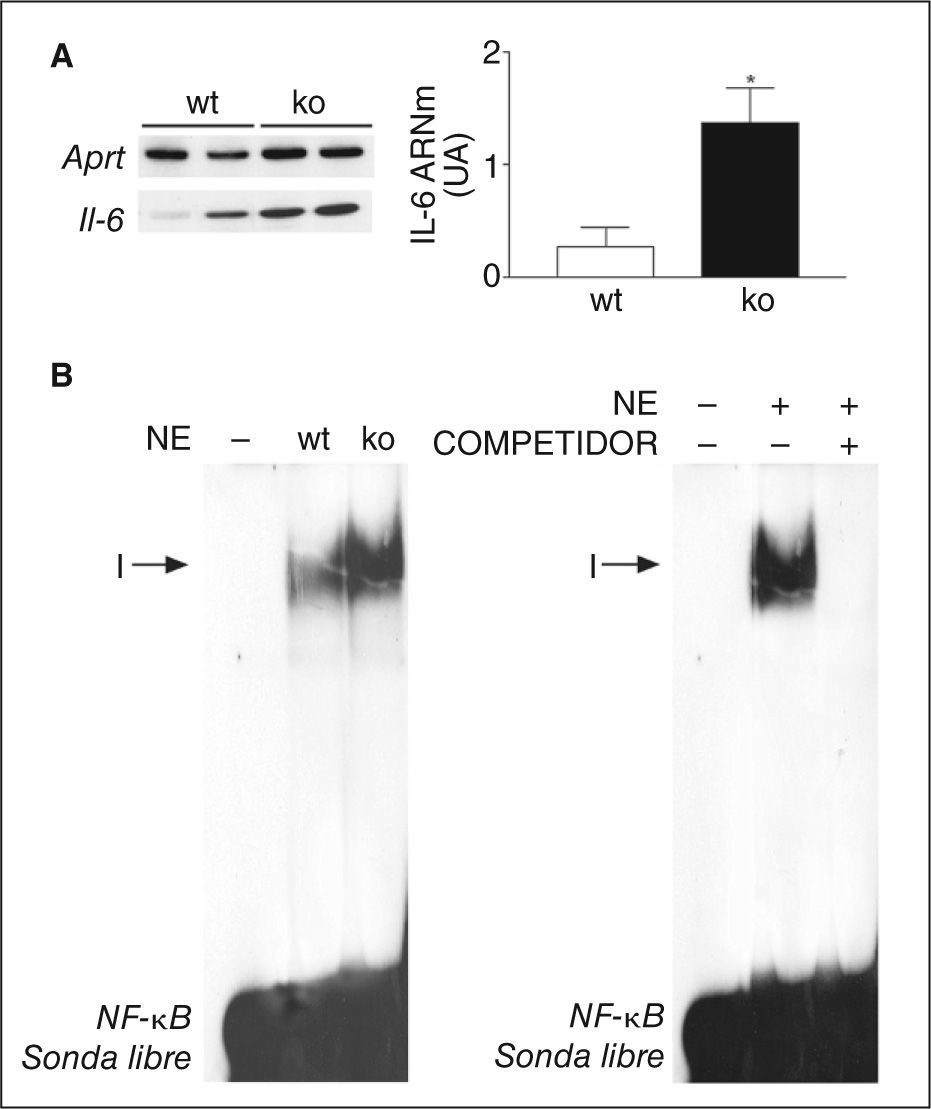
Los resultados anteriores indican que la activación de PPARβ/δ puede prevenir la producción de la IL-6 en adipocitos gracias a su capacidad para inhibir NF-κB. Para confirmar la capacidad de PPARβ/δ para inhibir NF-κB y la expresión de IL-6, utilizamos ratones deficientes para el gen PPARβ/δ (PPARβ/δ-/-). En el tejido adiposo de estos ratones, los valores de expresión de IL-6 estaban incrementados (inducción de 4 veces; p < 0,05) (fig. 3A) respecto a los ratones wild-type. Además, la actividad de unión a ADN de NF-κB también estaba incrementada en el tejido adiposo de estos animales al compararlos con los ratones wild-type (fig. 3B).
 presentan un incremento de la actividad factor nuclear kappa B (NF-κB) en tejido adiposo blanco. A. Análisis de los valores de ácido ribonucleico mensajero (ARNm) de interleucina (IL) 6 en tejido adiposo blanco en ratones wild-type (wt) o PPARβ/δ-/- (ko). La cuantificación de la autorradiografía representativa se encuentra normalizada para los valores de Aprt. Los datos se expresan como la media ± desviación estándar de 5 experimentos independientes. *p < 0,05 frente a wild-type. B. Autorradiografía representativa del ensayo de retardación de la movilidad electroforética realizado con sonda de NF-κB marcada con 32P y extractos de proteína nuclear donde se puede apreciar la formación de un único complejo específico (I). UA: unidades arbitrarias.")
Los ratones deficientes para el activado por proliferadores peroxisómicos β/δ (PPARβ/δ-/-) presentan un incremento de la actividad factor nuclear kappa B (NF-κB) en tejido adiposo blanco. A. Análisis de los valores de ácido ribonucleico mensajero (ARNm) de interleucina (IL) 6 en tejido adiposo blanco en ratones wild-type (wt) o PPARβ/δ-/- (ko). La cuantificación de la autorradiografía representativa se encuentra normalizada para los valores de Aprt. Los datos se expresan como la media ± desviación estándar de 5 experimentos independientes. *p < 0,05 frente a wild-type. B. Autorradiografía representativa del ensayo de retardación de la movilidad electroforética realizado con sonda de NF-κB marcada con 32P y extractos de proteína nuclear donde se puede apreciar la formación de un único complejo específico (I). UA: unidades arbitrarias.
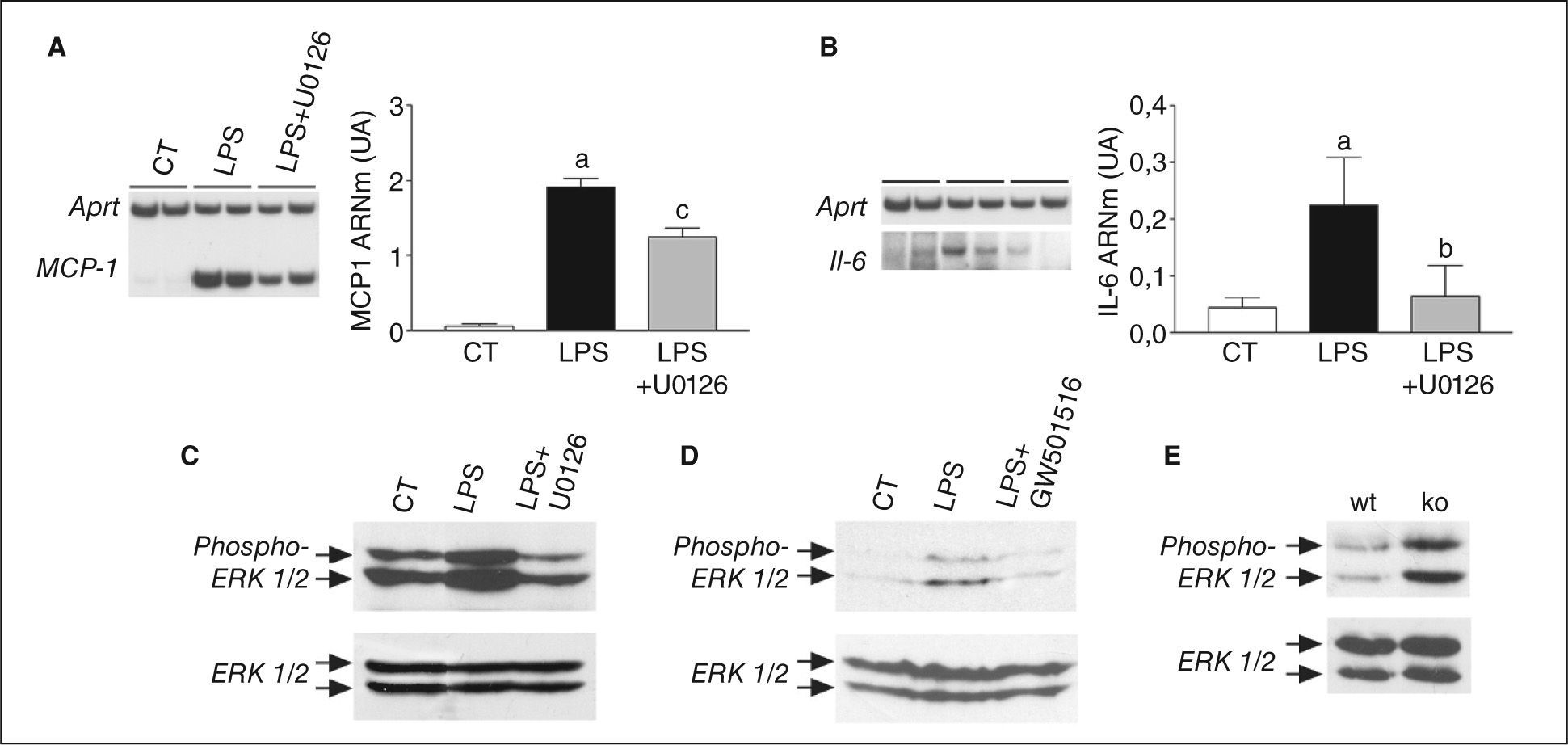
Seguidamente, intentamos determinar el mecanismo de la inhibición de NF-κB por PPARβ/δ. Puesto que recientemente se ha publicado que la activación de ERK1/2 induce la activación de NF-κB en adipocitos23, evaluamos la implicación de la cascada ERK-MAPK en la activación de NF-κB inducida por LPS. Para ello utilizamos U0126, un inhibidor específico de ERK1/2, que interacciona con MEK, inhibiendo la actividad catalítica de esta cinasa, y de este modo evita la fosforilación de ERK1/2. Las células estimuladas con LPS mostraron un incremento en los valores de ARNm de MCP-1 (inducción de 31 veces; p < 0,001) y de IL-6 (inducción de 4 veces; p < 0,05), mientras que el tratamiento con U0126 provocó reducciones del 35% (p < 0,001) y 72% (p < 0,05) respecto a la inducción observada en estos 2 genes por LPS, respectivamente (figs. 4A y B ). Como control, verificamos que el tratamiento con U0126 inhibía la activación de ERK1/2 provocada por LPS (fig. 4C).
 regula la fosforilación de ERK1/2 en adipocitos y tejido adiposo blanco. El inhibidor de MEK1/2, U0126, bloquea la activación del factor nuclear kappa B (NF-κB) inducida por lipopolisacárido (LPS). Efecto de U0126 sobre la expresión de Mcp-1 (A) e interleucina (IL) 6 (B) en adipocitos 3T3-L1. La cuantificación de las autorradiografías representativas se encuentran normalizadas para los valores de Aprt. Los datos se expresan como la media ± desviación estándar de 5 experimentos independientes. ap < 0,001 frente a control. bp < 0,001 y cp < 0,05 frente a células estimuladas con LPS. Análisis de ERK1/2 total y fosforilada por inmunodetección de extractos de proteína total de adipocitos 3T3-L1 estimulados con 100ng/ml de de LPS durante 10min en presencia o ausencia de 0,5μmol/l de GW501516 (C) o de U0126 (D), y en tejido adiposo blanco de ratones wild-type (wt) o PPARβ/δ-/- (ko) (E).")
El receptor activado por proliferadores β/δ (PPARβ/δ) regula la fosforilación de ERK1/2 en adipocitos y tejido adiposo blanco. El inhibidor de MEK1/2, U0126, bloquea la activación del factor nuclear kappa B (NF-κB) inducida por lipopolisacárido (LPS). Efecto de U0126 sobre la expresión de Mcp-1 (A) e interleucina (IL) 6 (B) en adipocitos 3T3-L1. La cuantificación de las autorradiografías representativas se encuentran normalizadas para los valores de Aprt. Los datos se expresan como la media ± desviación estándar de 5 experimentos independientes. ap < 0,001 frente a control. bp < 0,001 y cp < 0,05 frente a células estimuladas con LPS. Análisis de ERK1/2 total y fosforilada por inmunodetección de extractos de proteína total de adipocitos 3T3-L1 estimulados con 100ng/ml de de LPS durante 10min en presencia o ausencia de 0,5μmol/l de GW501516 (C) o de U0126 (D), y en tejido adiposo blanco de ratones wild-type (wt) o PPARβ/δ-/- (ko) (E).
Finalmente, examinamos si el ligando de PPARβ/δ evitaba la activación de NF-κB e inhibía la fosforilación de ERK1/2 en adipocitos. La inmunodetección de ERK1/2 total y fosforilada (fig. 4D) reveló que la activación de esta vía provocada por LPS no se producía en presencia del agonista de PPARβ/δ. Estos resultados indican que la activación de este receptor nuclear previene la activación de NF-κB a través de la inhibición de la fosforilación de ERK1/2. Para acabar de confirmar que PPARβ/δ regulaba la fosforilación de ERK1/2, determinamos la expresión de esta cinasa en tejido adiposo de ratones PPARβ/δ-/-. Estos ratones presentaban un incremento en la fosforilación de ERK1/2, en comparación con los ratones wild-type (fig. 4E), lo que indicaba la implicación de este receptor nuclear en la regulación de esta vía.
DiscusiónObesidad, resistencia a la insulina y diabetes mellitus tipo 2 son fenómenos íntimamente asociados a la presencia de un proceso inflamatorio crónico de intensidad baja, caracterizado por una producción anormal de citocinas24. En todo este proceso, los adipocitos desempeñan un papel fundamental, ya que son una de las principales fuentes de producción de citocinas (TNF-α, IL-6 o MCP-1). Estas citocinas se secretan como resultado de la activación de distintas cascadas de señalización implicadas en la resistencia a la insulina inducida por obesidad25. La activación crónica del factor de transcripción proinflamatorio NF-κB se ha descrito previamente como una de las principales cascadas de señalización que relaciona la inflamación con la obesidad y la diabetes mellitus tipo 226. La activación de este factor de transcripción puede deberse al aumento de los valores de moléculas, como los ácidos grasos o el LPS24. Distintos estudios han propuesto que el LPS participa iniciando la respuesta inflamatoria observada en obesidad y diabetes mellitus tipo 2. Además, los valores de LPS circulantes son mayores en pacientes diabéticos de tipo 2 que en individuos sanos, y el LPS estimula la secreción de citocinas proinflamatorias en adipocitos humanos27.
Los resultados obtenidos en este estudio indican que la activación de PPARβ/δ en adipocitos evita el incremento en la expresión y la secreción de citocinas inducido por LPS, un efecto probablemente debido a su capacidad para prevenir la activación de NF-κB. Estudios previos han propuesto que la activación de PPARβ/δ inhibe la activación de NF-κB en células cardíacas, a través de la interacción proteína-proteína entre PPARβ/δ y la subunidad p65 de NF-κB, o el incremento de los valores de la proteína represora IκBα28,29. En nuestras condiciones de trabajo no observamos cambios en la interacción PPARβ/δ-p65, ni en los valores de proteína IκBα en adipocitos, después del tratamiento con GW501516 (resultados no mostrados). Estos datos indican que el efecto antiinflamatorio de este fármaco en adipocitos es independiente de estos mecanismos. Por otro lado, estudios recientes indican que la activación de ERK1/2 está implicada en la activación del proceso inflamatorio en adipocitos26 mediante el incremento de la actividad de NF-κB23. De acuerdo con esta posibilidad, observamos una reducción en la expresión de citocinas proinflamatorias en adipocitos 3T3-L1 estimulados con LPS en presencia del inhibidor de esta vía U0126. Estos resultados concuerdan con estudios previos que han demostrado una reducción en la expresión de citocinas en adipocitos humanos en presencia de este inhibidor23. Además, nuestros resultados parecen demostrar que GW501516 puede prevenir la activación de NF-κB gracias a su capacidad para inhibir ERK1/2. Este hecho concuerda con el incremento en los valores de ERK1/2 fosforilada y el incremento de la actividad NF-κB encontrado en el tejido adiposo de ratones PPARβ/δ-/-. Esta regulación de ERK1/2 por parte de PPARβ/δ ya se había descrito en otros tipos celulares. Así, la activación de PPARβ/δ por GW0742 reduce los valores de ERK1/2 fosforilada en queratinocitos humanos30. Todos estos datos parecen confirmar que PPARβ/δ puede regular la fosforilación de ERK1/2 en distintos tejidos. Sin embargo, son necesarios nuevos estudios para investigar los mecanismos moleculares mediante de los cuales PPARβ/δ regula ERK1/2.
En resumen, los resultados obtenidos en este estudio parecen indicar que PPARβ/δ previene la activación por LPS de NF-κB a través de la inhibición de ERK1/2, con lo que así se reduce la producción de citocinas proinflamatorias implicadas en el desarrollo de la resistencia a la insulina.
Ricardo Rodríguez-Calvo es becario de la fundación Ramón Areces. Xavier Palomer es becario del Ministerio de Educación y Ciencia. Este estudio ha sido financiado por Swiss National Science Foundation, Generalitat de Catalunya (SGROS-00833), Fundación Ramón Areces, Ministerio de Educación y Ciencia (SAF-2006-01475), la Unión Europea y la Fundació Privada Catalana de Nutrició i Lípids. El CIBER de Diabetes y Enfermedades Metabólicas Asociadas es una iniciativa del ISCIII (Ministerio de Ciencia e Innovación).
Una comunicación referente a esta línea de trabajo, titulada “PPARβ/δ inhibe la inducción de citocinas en adipocitos mediante la prevención de la activación NF-κB a través de ERK1/2”, se presentó en el XXI Congreso Nacional de la SEA (Madrid 2008) y fue galardonada con una mención especial.