




In vitro ceramide-enriched LDL (CER-LDL) reproduces most of the properties of electronegative LDL (LDL(−)), a heterogeneous subfraction of LDL found in plasma. LDL(−) comprises several modifications of LDL and has an increased content in ceramide (CER). It promotes cytokine release in monocytes through CD14 and TLR4. CER-LDL also induces cytokine release in these cells but the mechanism is unknown.
AimTo evaluate TLR4 andCD14 as the putative receptors involved in cytokine release induced by CER-LDL.
MethodsCER-LDL was obtained by incubating native LDL with CER-enriched liposomes. CER content in CER-LDL was assessed by thin layer chromatography of lipid extracts. CER-LDL and LDL(−) were incubated for 20h with human monocytes in the presence or absence of a TLR4 signaling inhibitor. Both LDLs were also incubated with two human monocytic cell lines, normal and THP1 overexpressing CD14 (THP1-CD14) cells. The release of IL-6, IL-10 and MCP-1 was evaluated by ELISA in culture medium.
ResultsThe release of IL-6, IL-10 and MCP-1 induced by CER-LDL in monocytes was inhibited by VIPER (90% inhibition), a specific TLR4 inhibitor. The cytokine release induced by CER-LDL was negligible in THP1, cells presenting a very low CD14 expression. In contrast, the induction of cytokine release in THP1-CD14 was high and dependent on the CER content in LDL.
ConclusionCER-LDL induces IL-6, IL-10 and MCP-1 release through the activation of CD14 and TLR4 in monocytes, reproducing the behavior of LDL(−). The increased content of CER in LDL(−) is then related to the inflammatory action of LDL(−).
La LDL enriquecida in vitro en ceramida (CER-LDL) reproduce varias características atribuidas a la LDL electronegativa (LDL(−)), subfracción heterogénea de LDL presente en circulación que induce la liberación de citoquinas en monocitos mediante CD14 y TLR4. La CER-LDL estimula también la liberación de citoquinas en monocitos, aunque el mecanismo se desconoce.
ObjetivoEvaluar si CD14-TLR4 son receptores activados por la CER-LDL para inducir la liberación de citoquinas.
Material y métodosLa CER-LDL se obtuvo in vitro mediante incubación de LDL nativa con liposomas enriquecidos en CER. El contenido en CER de la CER-LDL fue evaluado mediante cromatografía en capa fina. La CER-LDL y la LDL(−) fueron incubadas 20h con monocitos humanos en presencia o ausencia de un inhibidor de la señalización de TLR4. También se incubaron con 2líneas de monocitos humanos, THP1 y THP1, que sobreexpresan CD14 (THP1-CD14). Se evaluaron IL-6, IL-10 y MCP-1 en todos los sobrenadantes celulares mediante ELISA.
ResultadosLa liberación de IL-6, IL-10 y MCP-1 inducida por la CER-LDL en monocitos fue inhibida mediante VIPER (90% de inhibición), inhibidor específico de TLR4. Las citoquinas liberadas por la CER-LDL fueron escasas en THP1, células que presentan baja expresión de CD14. Las citoquinas liberadas por la CER-LDL en THP1-CD14 fueron superiores y dependientes del contenido en CER de la LDL.
ConclusiónLa LDL-CER induce IL-6, IL-10 y MCP-1 a través de la activación de CD14-TLR4 en monocitos, mimetizando a la LDL(−). La acción inflamatoria de la LDL(−) está relacionada con su contenido aumentado en CER.
Atherosclerosis is a chronic progressive disease caused by the accumulation of lipids and fibrous elements in preferred lesion sites of the large arteries.1 Lipoproteins, and particularly low-density lipoproteins (LDL), are involved in atherosclerosis, as they not only pass through the endothelial cells and accumulate in the fatty streak, but also stimulate the expression of inflammatory molecules in endothelium and other cells of the immune system.2 Native LDL does not present these properties, but it becomes proatherogenic when it is modified. Most LDL modifications found in vivo take place when LDL is trapped in the subendothelial space by elements of the extracellular matrix, such as proteoglycans.3,4
The most widely studied modification of LDL is oxidation, caused by oxidative enzymes, such as myeloperoxidase and lipooxygenase, and by reactive oxygen species secreted by cells of the artery wall into the subendothelial space.5,6 Oxidized LDL has inflammatory properties.7 LDL can also be modified by other enzymes such as hepatic lipase, cholesterol esterase and numerous phospholipases. These modifications yield high atherogenicity.8 Most of these modifications promote changes in the structure and composition of LDL, rendering this lipoprotein to aggregation or particle fusion.9 Another common feature of these modified forms of LDL is the high electronegative charge, compared to native LDL.10
Modified LDL can be generated not only in the subendothelial space but also while it is circulating in blood, where oxidized lipids from the diet, hydroperoxides from cell membranes and several hydrolytic enzymes are present.4,11 Avogaro et al. were the first to describe the presence of a heterogeneous modified LDL in circulation, called electronegative LDL (LDL(−)).12 Although LDL(−) represents less than 5% of the total LDL in normolipemic subjects, it is increased in patients with disorders associated with a higher cardiovascular risk, such as hypercholesterolemia13 or type I and II diabetes mellitus.14
LDL(−) differs from native LDL (LDL(+)) on its physicochemical characteristics. It has a different size and density distribution, a different lipid and protein composition and altered apoB-100 conformation (apoB).15 LDL(−) display phospholipase activity, such as platelet associated factor acetyl-hydrolase (PAF-AH),16 which degrades PAF-like phospholipids, and phospholipase C (PLC)-like activity.17 PLC-like activity degrades choline-containing phospholipids yielding ceramide (CER), monoacylglycerol (MAG) and diacylglycerol (DAG). The changes in lipid content induced by PLC-like activity in LDL(−) modify the surface structure of the particles9 and they have been related to a higher tendency to aggregation and proteoglycan binding of LDL(−).18
LDL(−) promotes cytotoxicity, apoptosis and inflammation.19 The induction of cytokine release by LDL(−) was first described in endothelial cells,20 LDL(−) also induces the release of inflammatory mediators such as IL-6, IL-10 and MCP-1 in monocytes and lymphocytes by promoting gene transcription.21,22 Our group reported that receptors involved in cytokine release promoted by LDL(−) in monocytes are TLR4 and CD14.23 These receptors are typically from innate immune response and recognize lipopolysaccharide (LPS) present in the bacterial wall. For this reason, LDL(−) and LPS compete in the inflammatory effect promoted in monocytes.23
CER is not only an essential component of cell membranes but also is an inflammatory molecule that can regulate vital cell functions.24 Some mediators of the acute phase response such as LPS and inflammatory cytokines induce CER formation in liver due to activation of sphingomyelinase. CER is then secreted into the bloodstream as a component of LDL and VLDL25 and subsequently accumulates in atherosclerotic lesions.26 It has been shown that, as a consequence of the degradation of SM by its PLC-like activity, LDL(−) has an increased content in CER.27 This content is, in part, responsible for the cytokine release promoted by this lipoprotein in monocytes. HDL, an atheroprotective lipoprotein found to diminish the cytokine release induced by LDL(−),28 is also capable of reducing the CER content in LDL(−).27 Moreover, CER-enriched LDL (CER-LDL) reproduces some properties ascribed to LDL(−), including the ability to induce IL-6, IL-10 and MCP-1 release in monocytes.27 It has been reported that CD14 recognizes CER.25,29
The aim of this study was then to assess whether CER-LDL induces cytokine release through CD14-TLR4 pathway.
MethodsLDL subfraction isolationPlasma samples from a pool of healthy normolipemic subjects (total cholesterol<5.2mmol/L, triglyceride<1mmol/L) were obtained in EDTA-containing Vacutainer tubes. All subjects gave their written informed consent and the study was conducted after approval from the hospital ethics committee. LDL (1.019–1.050g/mL) was isolated by sequential flotation ultracentrifugation (20h at 36,000rpm, 4°C). Total LDL was subfractionated in LDL(+) and LDL(−) by preparative anion-exchange chromatography in an ÄKTA-Fast protein liquid chromatography (FPLC) system (GE Healthcare) as described.28
Enrichment of LDL with ceramideLDL was enriched with N-acetyl-d-sphingosine (ceramide, CER) (Sigma Aldrich) by means of liposomes enriched with this compound, according Boyanovsky et al.30 Briefly, liposomes were formed by extracting lipids from native LDL (250μg) according to the Bligh and Dyer method,31 followed by incubation with CER (5 and 10μM). Dried lipid extracts were reconstituted in KBr solution and sonicated. Liposomes, used as donor vesicles, were incubated with LDL (acceptor molecule) at 37°C for 45min. LDL incubated with CER non-enriched liposomes, termed as CER-LDL 0μM, was processed in parallel. LDL was re-isolated from liposomes by ultracentrifugation.
Lipid compositionCholesterol, trygliceride, apoB (Roche Diagnostics), phospholipid and NEFA (Wako chemicals) content in CER-LDL samples was determined in a Cobas c501 autoanalyser. Results were expressed as the percentage of lipoprotein mass for all the components except NEFA, which was stated as molNEFA/mol apoB.
Thin layer chromatography (TLC)The enrichment of CER in LDL was tested after lipid extraction (250μg apoB).31 Lipid extracts were reconstituted with 20μl chloroform and applied to TLC silica gel plates. Three sequential mobile phases were used to develop the plates: phase 1, chloroform/methanol/water (65:40:5, v/v/v) to 5cm; phase 2, toluene/diethilether/ethanol (60:40:3, v/v/v) to 13cm; phase 3, heptane to 17cm. Lipids were stained in ethanol containing 5% phosphomolibdic acid and 5% sulphuric acid and the plate was dried for 10min at 100°C. Lipid dots were detected in a ChemiDoc 2000 (Biorad).27
Primary monocytes and THP1 cells culturePrimary monocytes were isolated from peripheral blood of human volunteers. This procedure was approved by the institutional ethics committee and subjects gave their written informed consent. Cells were isolated according to their density22,32 by gradient centrifugation (Lympholyte, Cedarlane). Differentiation of monocytes and lymphocytes was performed depending on their adhesive properties after 4h of culture. The purity of monocytes was assessed by flow-cytometry with a four marker combination with a four-marker combination, and by May Grünwald/Giemsa staining (monocyte purity 80–85%). Cell viability was verified by LDH assay (Roche Diagnostics, Basel, Switzerland). Cells were cultured in 12-well plates (106 cells/well) with RPMI 1640 medium with 10% fetal calf serum 24h before the addition of CER-LDL (150mgapoB/L).
THP1-Xblue™ (THP1) cells and THP1-Xblue™-MD2-CD14 (THP1-CD14) (Invivogen) derived from human monocytic THP1 were also used. Both cell lines naturally express TLRs, but THP1-Xblue™-MD2-CD14 overexpresses MD2 and CD14 increasing the response to TLR ligands. Cells were cultured in 12-well plates (106cells/well) in RPMI supplemented with Normocin (100mg/L) and Zeocin (200mg/L) for THP1-XBlue™ cells, and Zeocin (200mg/L) and G418 (250mg/L) for THP1-XBlue™-MD2-CD14. Cells were seeded 24h before the addition of CER-LDL (150mg apoB/L).
Cytokine release experimentsCER-LDLs, previously dialyzed against RPMI 1640 medium and filtered in sterile conditions, were added at 150mg apoB/L to primary monocytes, THP1 or THP1-CD14. The incubation of cells with LDLs was performed in RPMI deficient medium (1% fetal calf serum). The cell supernatant was collected after 20h incubation and the concentration of IL-6, IL-10 and MCP-1 released to the culture medium was quantified by ELISA (Bender Medsystems), as described.22
The TLR4 pathway in primary monocytes was inhibited with the Viral Inhibitor Peptide (VIPER) (Imgenex), which binds to TIR domains of TLR4 and the adaptor proteins Mal and TRAM, blocking the interaction between them. Cells were treated with 30μM VIPER for 2h before adding CER-LDLs. CP7 peptide (Imgenex) was used as a negative control. Both compounds were reconstituted following the manufacturer's instructions.
StatisticsData were analyzed using SPSS 11.5 for Windows. Results were expressed as mean±SD. Samples were considered as paired data and Mann–Whitney assay test was done.
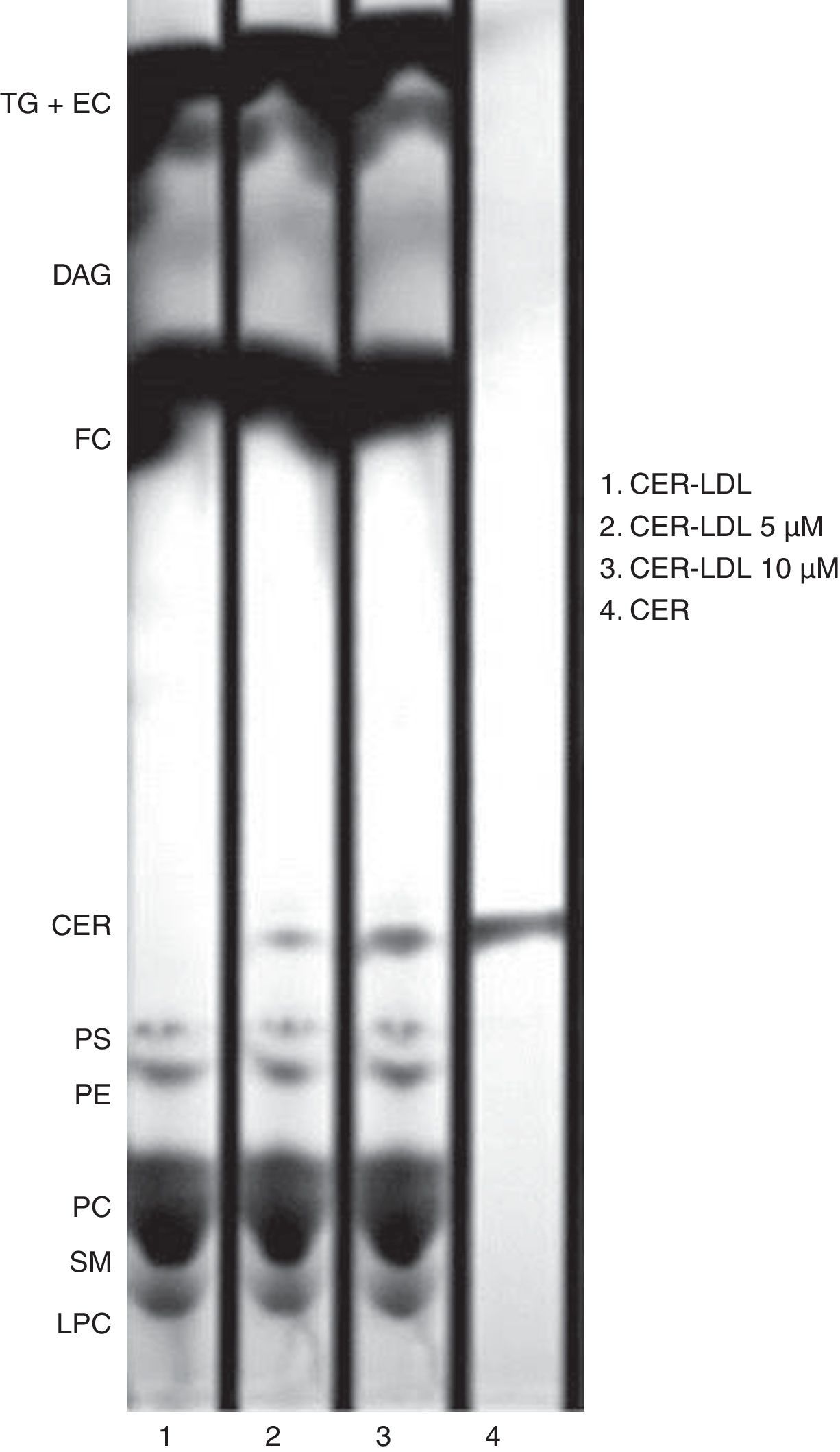
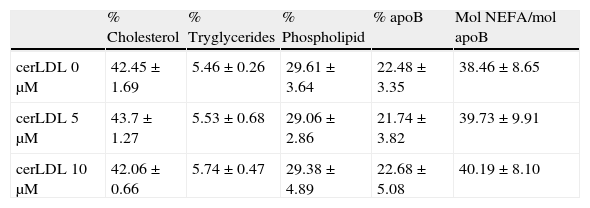
ResultsEnrichment of LDL in CERThe enrichment of LDL in ceramide was tested by TLC. CER-LDL 5μM and 10μM increased its content in CER in a dose-dependent manner in comparison with CER-LDL 0μM. We did not observe any other changes in the phospholipid content (Fig. 1), cholesterol, tryglicerides, phospholipids, NEFA or apoB content in CER-LDL (Table 1).

Representative TLC of lipid extracts of CER-LDL 0, 5 and 10μM. CER-LDL 0μM was LDL processed in parallel but with no addition of CER. The CER used as compound was added to the TLC silica gel plate as standard. TG: triglycerides, EC: esterified cholesterol, DAG: diacylglycerol, FC: free cholesterol, CER: ceramide, PS: phosphatidylserine, PE: phosphatidylethanolamine, PC: phosphatidylcholine, SM: sphigomyelin, LPC: lisophosphatidylcholine.
Cholesterol, tryglycerides, phospholipids, apoB and NEFA composition of CER-LDL 0, 5 and 10μM (n=5).
| % Cholesterol | % Tryglycerides | % Phospholipid | % apoB | Mol NEFA/mol apoB | |
| cerLDL 0μM | 42.45±1.69 | 5.46±0.26 | 29.61±3.64 | 22.48±3.35 | 38.46±8.65 |
| cerLDL 5μM | 43.7±1.27 | 5.53±0.68 | 29.06±2.86 | 21.74±3.82 | 39.73±9.91 |
| cerLDL 10μM | 42.06±0.66 | 5.74±0.47 | 29.38±4.89 | 22.68±5.08 | 40.19±8.10 |
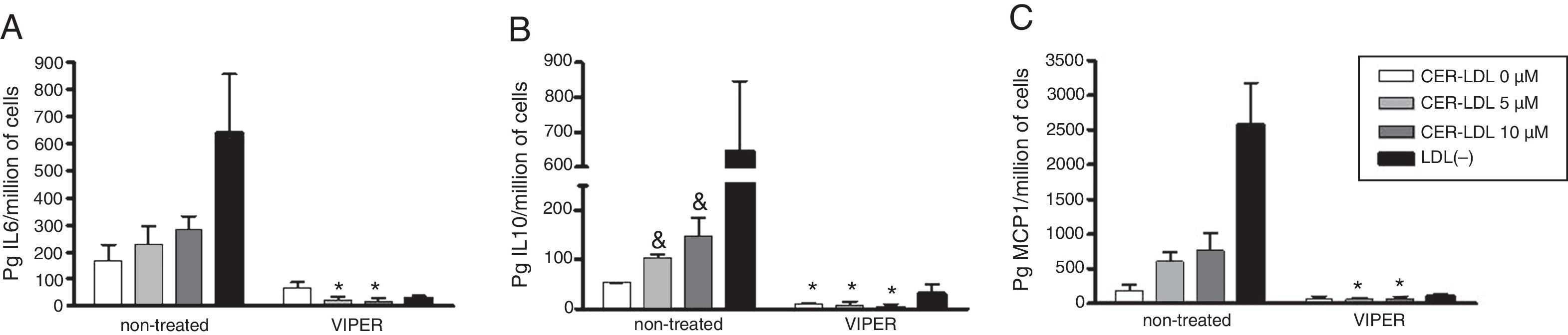
The involvement of TLR4 in the inflammatory action of CER-LDL was tested by the treatment of primary monocytes with VIPER compared to non-treated cells (Fig. 2). As described,27 the addition of CER-LDL 5 and 10μM produced a dose-dependent elevation of IL-6 (2A), IL-10 (2B) and MCP-1 (2C) on monocytes non-treated with VIPER. In contrast, IL-6, IL-10 and MCP-1 induced by CER-LDL in monocytes treated with VIPER were significantly lower than in non-treated cells, reaching inhibitions of 90%. There was no difference between CER-LDL 5 and 10μM in the presence of VIPER in any of the cytokines analyzed. LDL(−) was used as a positive control of TLR4-mediated inflammatory effect. In non-treated monocytes, cytokine release promoted by LDL(−) was higher than that induced by CER-LDL 10μM in all cases.
, IL-10 (B) and MCP-1 (C) release induced by CER-LDL 0, 5 and 10μM in primary monocytes previously treated or non-treated with VIPER (TLR4 pathway inhibitor). LDL(−) was used as a positive control. Results of n=4 experiments are expressed as mean±SD, *treated with VIPER vs non-treated, &CER-LDL 5μM or 10μM vs 0μM.")
IL-6 (A), IL-10 (B) and MCP-1 (C) release induced by CER-LDL 0, 5 and 10μM in primary monocytes previously treated or non-treated with VIPER (TLR4 pathway inhibitor). LDL(−) was used as a positive control. Results of n=4 experiments are expressed as mean±SD, *treated with VIPER vs non-treated, &CER-LDL 5μM or 10μM vs 0μM.
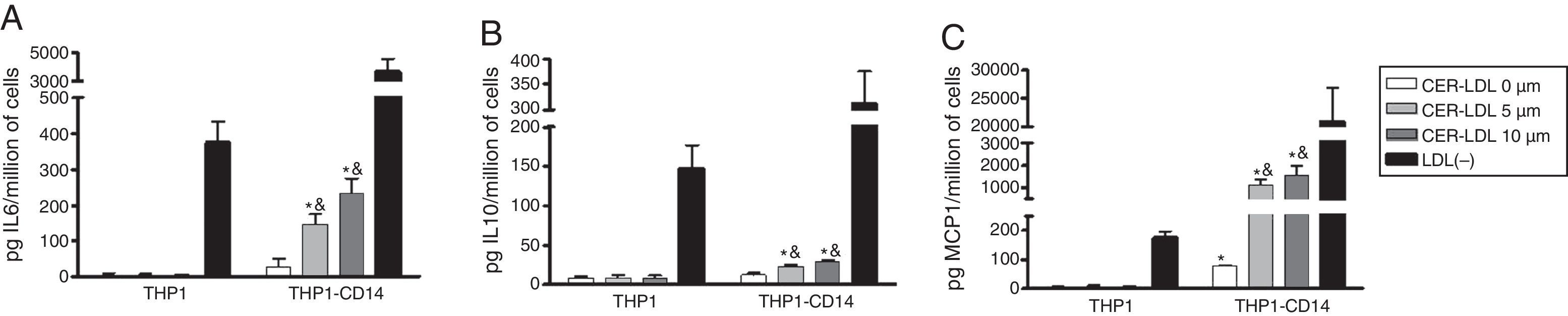
The involvement of CD14 on the cytokine release promoted by CER-LDL was tested in THP1 cells that overexpress CD14 and MD2. CER-LDL induced a minimal amount of cytokine release in THP1 cells (Fig. 3, left bars). However, CER-LDL induced IL-6 (3A), IL-10 (3B) and MCP-1 (3C) release in THP1-CD14 in a dose-dependent manner. In contrast to CER-LDL, LDL(−) induced a moderate cytokine release in THP1 cells, which was strongly increased in THP1-CD14 cells. The cytokine release induced by LDL(−) in THP1-CD14 was also higher than that induced by CER-LDL 10μM.
Discussion, IL-10 (B) and MCP-1 (C) release induced by CER-LDL 0, 5 and 10μM in THP1 and THP1 overexpressing MD2-CD14 (THP1-CD14). LDL(−) was used as a positive control. Results of n=4 experiments are expressed as mean±SD, *THP1-CD14 vs THP1, &CER-LDL 5μM or 10μM vs 0μM.")
This study shows that CER-LDL induces cytokine release in human primary monocytes through CD14 and TLR4, which is the same pathway activated by LDL(−). Previous findings reported that CER-LDL shares other common properties with LDL(−).27 These observations suggest that the increased CER content in LDL(−) is involved in some of the atherogenic properties of this LDL subfraction.
The role of CER and CER-LDL in inflammation and atherogenesis is supported by several observations. CER participates in the regulation of cell proliferation and differentiation, inflammation and apoptosis.33 Moreover, a relationship between increased CER content in LDL and the development of inflammatory diseases has been described.25 Retained LDL in atherosclerotic lesions accumulates CER.26 In macrophages, foam cell formation is induced by the promotion of CER-LDL uptake.34 It has recently been suggested that CER-LDL mimics some of the inflammatory effects of LDL(−).27 Such data and the current findings support a relationship between the increased CER content in LDL(−) and its inflammatory action in monocytes which is mediated through CD14-TLR4.
CD14 and TLR4 are innate pattern recognition receptors known to play a key role in initiating inflammatory responses to pathogen-associated molecular patterns, such as LPS.35 LPS promotes inflammation in two steps: first, LPS binds to CD14, which in turn associates with TLR4, thereby initiating the intracellular signaling cascade leading to cytokine release. Some modified LDLs, such as an early type of oxidized LDL (mmLDL), can also induce the CD14-TLR4 pathway.36 Recently, we reported that LDL(−) also activates CD14-TLR4 leading to IL-6, IL-10 and MCP-1 release in primary monocytes.23 In the current study we found that CER-LDL also induces cytokine release by the same pathway. In human primary monocytes, the addition of VIPER, a specific TLR4 pathway inhibitor, produced a significant fall in the release of IL-6, IL-10 and MCP-1 induced by CER-LDL, demonstrating the importance of TLR4 in mediating the inflammatory effects of CER-LDL. The presence of CD14 is also essential in the cytokine release promoted by CER-LDL, since the cytokine induction in a monocytic cell line lacking CD14 was almost undetectable. In contrast, the same monocyte line overexpressing CD14 released cytokines in a dose-dependent manner in response to CER-LDL. It had been described that CER can bind to CD14.29 Modification of LDL by enrichment with CER may change the surface structure of LDL,9 making it more available to be recognized by CD14. As a result, CD14 would detect CER from CER-LDL and then would complex with TLR4, inducing intracellular signals that lead to the inflammatory effect. Such behavior is similar to that described for LDL(−), although some differences exist. CD14 binds LDL(−) but TLR4 can also recognize this modified LDL23; hence, cytokine induction continues in the absence of CD14, although to a much lower extent than in the presence of CD14.23 CER-LDL induces cytokine release in a milder manner than LDL(−). It concurs with previous studies suggesting that LDL(−) must have inflammatory components other than CER.27 NEFA content is increased in LDL(−) and can also bind to CD14.37 NEFA have been related to the cytokine release promoted by LDL(−) in monocytes28 and in endothelial cells.38 In endothelial cells, LPC is also involved in IL-8 and MCP-1 release.38 CER metabolites such as sphingosine (SPH) and sphingosine-1-phosphate (S1P) might also be responsible for the inflammatory effect promoted by LDL(−). CER can be hydrolyzed by ceramidases to generate SPH, which can be phosphorylated to sphingosine-1-phosphate (S1P).39 CER, SPH and S1P are bioactive lipids enrolled in atherogenic processes in that they can accumulate in atherosclerotic lesions and participate in the regulation of signal transduction pathways.24 S1P promotes monocyte attachment and migration, proliferation of smooth muscle cells and activation of the NF-κB pathway, leading to inflammatory cytokines production.40 Further investigations are needed to ascertain all the components involved in LDL(−) inflammatory action and their relative role.
In conclusion, this work provides new insights into the mechanism leading to the cytokine release promoted by CER-LDL and LDL(−) in monocytes. We showed that the in vitro modification of LDL by enrichment with CER induces cytokine release through TLR4-CD14 in monocytes. CER-LDL may bind to CD14 forming a complex with TLR4 to initiate the intracellular pathways leading to cytokine release. The similarities of CER-LDL with LDL(−) suggest that the increased content in CER is responsible for part of the inflammatory effect of LDL(−).
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicts of interestThe author has no conflicts of interest to declare.
This work was supported by Grants from the Instituto de Salud Carlos III/Fondo de Investigación Sanitaria FIS PI 09/0160, PI012/967, PI10/0265 and RD12/0042/0043. All the authors are members of the 2009-SGR-1205 Research Group of the Generalitat de Catalunya.
A communication regarding this subject, entitled: “The HDL-induced inhibition of the ceramide content in the electronegative LDL is related to its inflammatory effect” was presented at XXV National Congress of the Spanish Society of Atherosclerosis (SEA) in Reus 2012 and awarded Special Mention (Ref ME/2012-21.30).