





La enfermedad del hígado graso no alcohólico cursa, en sus fases iniciales, con hipertrigliceridemia y acúmulo de lípidos en el hígado (esteatosis hepática). El ácido bempedoico es un inhibidor de la ATP:citrato liasa que promueve una inhibición dual de la síntesis de colesterol y ácidos grasos. Sin embargo, no se ha investigado su efecto en la prevención/tratamiento de la esteatosis hepática y la hipertrigliceridemia. El objetivo de nuestro trabajo ha sido elucidar si el ácido bempedoico, mediante un mecanismo diferente/alternativo a la inhibición de la ATP:citrato liasa, revierte estas alteraciones metabólicas.
Diseño experimentalEl estudio se realizó con un modelo animal de rata Sprague-Dawley hembra alimentada, durante 3 meses, con una dieta rica en grasa saturada suplementada con fructosa al 10% (p/v) en el agua de bebida. Se administró, durante el último mes, ácido bempedoico (30mg/kg/día) a un grupo de animales. Se analizaron parámetros zoométricos, se realizaron valoraciones plasmáticas, de expresión génica y proteica en muestras de hígado y se determinó la actividad de unión PPAR-PPRE.
ResultadosNuestro modelo de intervención dietética desarrolló esteatosis hepática e hipertrigliceridemia. A pesar de un aumento en la ingesta calórica total, no se observó un incremento de peso corporal de los animales. La administración de ácido bempedoico redujo significativamente la esteatosis hepática y promovió una marcada hipertrofia de los hepatocitos. Se observó un incremento del 66% en el peso del hígado de los animales tratados con el fármaco, que no se acompañó de modificaciones en los marcadores de inflamación, estrés oxidativo o estrés de retículo endoplasmático. El ácido bempedoico activó el receptor nuclear activado por proliferadores peroxisómicos (PPARα) y sus genes diana.
ConclusionesEl ácido bempedoico podría ser una terapia efectiva para el tratamiento del hígado graso y el riesgo cardiovascular asociado. El ácido bempedoico presenta otros mecanismos de acción diferentes a la inhibición de la ATP:citrato liasa, como sería la activación de PPARα, lo que podría explicar la reducción de la esteatosis hepática y el incremento del peso del hígado observado en los animales tratados con el fármaco⋅
In its initial stages, nonalcoholic fatty liver disease presents hypertriglyceridemia and accumulation of lipids in the liver (hepatic steatosis). Bempedoic acid is an ATP:citrate lyase inhibitor that promotes a dual inhibition of the synthesis of cholesterol and fatty acids. However, its effect in the prevention / treatment of hepatic steatosis and hypertriglyceridemia has not been investigated. The aim of our work has been to elucidate whether bempedoic acid, through a mechanism other than ATP:citrate lyase inhibition, reverses these metabolic alterations.
Experimental designThe study was carried out in female Sprague-Dawley rats fed, for three months, with a high fat diet supplemented with fructose (10% w/v) in drinking water. During the last month, bempedoic acid (30mg/kg/day) was administered to a group of animals. Zoometric and plasmatic parameters were analyzed, gene and protein expression analysis were performed in liver samples and PPAR-PPRE binding activity was determined.
ResultsOur interventional model developed hepatic steatosis and hypertriglyceridemia. Despite an increase in total caloric intake, there was no increase in body weight of the animals. The administration of bempedoic acid significantly reduced hepatic steatosis and promoted a marked hepatocyte hypertrophy. There was a 66% increase in the liver weight of the animals treated with the drug that was not accompanied by modifications in the markers of inflammation, oxidative stress, or endoplasmic reticulum stress. Bempedoic acid activated the peroxisome proliferator activated nuclear receptor (PPARα) and its target genes.
ConclusionsBempedoic acid could be an effective therapy for the treatment of fatty liver and associated cardiovascular risk. Bempedoic acid has other mechanisms of action besides the inhibition of ATP: citrate lyase, such as the activation of PPARα, which could explain the reduction in hepatic steatosis and the increase in liver weight observed in animals treated with the drug.
La enfermedad del hígado graso no alcohólico (non-alcoholic fatty liver disease, NAFLD) es una patología cuya incidencia se ha incrementado en las últimas décadas situándose su prevalencia actual por encima del 25%1. La NAFLD se caracteriza por presentar alteraciones derivadas, principalmente, de un estado de resistencia hepática a la insulina y englobadas dentro de las manifestaciones del síndrome metabólico y que se asocian a un incremento del riesgo cardiovascular2. Aunque la etiología de la NAFLD es múltiple, el consumo de una dieta tipo «western», con un elevado aporte calórico derivado de un alto contenido en azúcares simples y grasas saturadas juega un papel decisivo en su aparición3. Este tipo de dieta, sumado al sedentarismo, conduce a la obesidad y a la resistencia a la insulina (RI), dos factores etiopatogénicos clave en el desarrollo de NAFLD. La RI induce un mayor influjo de ácidos grasos (AG) hacia el hígado, provenientes del tejido adiposo4 y una activación hepática de la lipogénesis de novo (DNL)5,6 lo que comporta un aumento en la síntesis de triglicéridos hepáticos que, en un principio, serán exportados hacia el plasma, incorporados en las lipoproteínas de muy baja densidad (VLDL). La sobreproducción de VLDL en individuos con NAFLD implica dislipemia caracterizada por niveles elevados de triglicéridos y bajos de colesterol-HDL, además de un aumento de las partículas LDL pequeñas y densas, la tríada característica de la denominada dislipemia aterogénica7,8.
Aproximadamente, el 25% de los pacientes que presentan NAFLD acaban desarrollando esteatohepatitis no alcohólica o NASH que puede conllevar la aparición de fibrosis hepática y cirrosis9. En la actualidad, se están desarrollando fármacos moduladores selectivos del receptor PPARα, como el pemafibrato10, agonistas PPARα/δ como el elafibranor11 o inhibidores de acetil-CoA carboxilasa (ACC) como el GS-0976 o firsocostat12 para el tratamiento de NASH. Sin embargo, la acumulación de triglicéridos en el hígado (esteatosis hepática), que se considera una alteración que aparece en las primeras fases de la NAFLD, no tiene ningún tratamiento farmacológico plenamente eficaz en nuestros días. En la búsqueda de nuevas dianas farmacológicas, la ATP-citrato liasa (ACL) resulta de gran interés, puesto que es un enzima con funciones clave en la intersección entre el catabolismo de nutrientes y la biosíntesis de colesterol y ácidos grasos13. El ácido bempedoico (ABem; ETC-1002, ácido 8-hidroxi-2,2,14,14-tetrametilpentadecanodioico) es un activador de la AMP-proteina kinasa (AMPK) y un inhibidor de la ACL, cuyos efectos se han estudiado en modelos in vivo como inhibidor dual de la síntesis de colesterol y ácidos grasos14,15 y su comercialización ha sido aprobada recientemente por la Agencia Europea del Medicamento (EMA). Estudios clínicos en pacientes con hipercolesterolemia realizados por Ballantyne et al.16 mostraron que el ABem únicamente reducía los niveles de colesterol plasmático, pero no los de triglicéridos, a diferencia de lo observado en roedores17, probablemente debido a la escasa relevancia de la DNL en ausencia de resistencia a la insulina. Sin embargo, no se ha investigado el efecto del ABem en la prevención o el tratamiento de la esteatosis hepática y la hipertrigliceridemia asociada a NAFLD.
A partir de nuestra experiencia en el estudio de los efectos del consumo de fructosa líquida al 10% (p/v) sobre el metabolismo lipídico18,19, hemos diseñado un modelo dietético de hígado graso sin inflamación en rata Sprague-Dawley hembra alimentada con una dieta rica en ácidos grasos saturados (palmítico y esteárico), desprovista de colesterol y suplementada con fructosa líquida. El objetivo del presente trabajo ha sido estudiar si el ácido bempedoico podría constituir una alternativa terapéutica para el tratamiento de la esteatosis hepática y/o la hipertrigliceridemia, observada en las fases iniciales de NAFLD, y elucidar el mecanismo implicado.
Material y métodosDiseño experimentalPara este estudio se utilizaron 24 ratas hembra Sprague-Dawley que fueron suministradas por Charles River (Barcelona, España). Los animales se mantuvieron con agua y comida ad libitum, a temperatura y humedad constante con un ciclo de luz/oscuridad de 12horas durante una semana. Tras aclimatación, se distribuyeron aleatoriamente las ratas en tres grupos de estudio (n=8): un grupo control (CT) alimentado con una dieta estándar y agua de bebida, un grupo alimentado con una dieta rica en grasa (dieta high-fat: Teklad Custom Diet TD.180456 de Envigo, España). El contenido de esta dieta en %kcal es: proteínas 15,5%, carbohidratos 37,5% y grasas 47%, de las cuales 210g/kg son de mantequilla de cacao que contiene, aproximadamente, un 25% de ácido palmítico y un 35% de ácido esteárico20. La dieta se suplementó con fructosa en forma de solución al 10% (peso/volumen) en el agua de bebida (HF-FR). Un tercer grupo de ratas fue alimentado con HF-FR y recibió, el último mes, una dosis diaria de ácido bempedoico por vía oral de 30mg/kg, diluido en 0,5% de carboximetilcelulosa y 0,025% de tween-20 (ABem). Al resto de grupos se les administró el volumen correspondiente de vehículo del fármaco. Se controló la ingesta de comida y bebida cada 2 días y se pesaron los animales una vez a la semana. Tras 3 meses de estudio, las ratas se sacrificaron por exanguinación bajo anestesia con ketamina:xilacina (proporción 9:1) con un ayuno previo sacrificio de 2horas.
Todo el proceso se realizó de acuerdo con las directrices establecidas por el Comité de Bioética de la Universitat de Barcelona, según se indica en la Ley (5/1995) (21 de julio) de la Generalitat de Catalunya (número de expediente: 10106).
Obtención de muestrasPara la determinación de los niveles de triglicéridos y colesterol plasmáticos se obtuvieron muestras de sangre mediante punción de la vena safena. Para los niveles séricos de ácidos grasos libres (AGL), AST y ALT, la sangre se recolectó mediante punción cardíaca en el momento del sacrificio de los animales, en tubos Sarstedt AG & Co. (Nümbrecht, Alemania), y el suero se obtuvo por centrifugación a 1000×G durante 10minutos a temperatura ambiente. Los hígados de las ratas se extrajeron, se pesaron y se congelaron en N2 líquido de forma inmediata y se conservaron a −80°C hasta su utilización. 10-100mg de tejido hepático se usaron para la extracción del ARN total. Dos muestras adicionales de 250mg, aproximadamente, se utilizaron para la cuantificación de los lípidos hepáticos, para la obtención de proteína total y de extractos nucleares. Un fragmento de tejido hepático, antes de su congelación, se conservó en formaldehído para los estudios histológicos.
Determinación de lípidos plasmáticos y hepáticosLos niveles plasmáticos de triglicéridos y colesterol se determinaron mediante glucómetro Accutrend® Plus System glucometer (Cobas, RocheFarma, Barcelona) usando tiras reactivas específicas. Los ácidos grasos libres (AGL) se midieron mediante el ensayo colorimétrico de Bioo Scientific (Austin, TX, EE.UU.) y los niveles de AST y ALT mediante kits enzimáticos de Spinreact (Girona, España).
Los lípidos hepáticos se extrajeron básicamente siguiendo el protocolo descrito por Qu et al.21 40mg de hígado se homogenizaron en 800μl de acetona, usando un homogeneizador mecánico (Polytron® PT 1200E, Selecta). Tras una incubación de 12horas en agitación constante y a temperatura ambiente, se dejaron reposar las muestras 15minutos y se procedió a la valoración de los niveles de triglicéridos y colesterol mediante kits colorimétricos: 41030 Triglycerides y MD41021 Cholesterol, de Spinreact (Girona, España).
Estudios histológicosLas muestras de tejido hepático se deshidrataron y se incrustaron en parafina mediante un procesador automático de tejidos Leica TP1020. El bloque se formó en el centro de incrustación de tejidos EG1140H y las muestras se cortaron a 5micras en el PFM Rotary 3004M. La tinción con hematoxilina-eosina se realizó de forma automática en un termoshandon varistain 24-4. Las imágenes se adquirieron con un microscopio Leica DMSL equipado con una cámara DP72. La cuantificación de la hipertrofia se realizó mediante el programa informático ImageJ y se expresa en % de superficie ocupada por núcleos de hepatocitos por área de imagen. Todos los procedimientos se realizaron en el laboratorio de Histopatología Animal de la Universitat de Barcelona.
Análisis del ARN: RT-qPCREl ARN total se aisló a partir de 100mg de tejido hepático y mediante el reactivo TRIsureTM (Meridian Biosciences, Menfis, TN, EUA), de acuerdo con las instrucciones del fabricante. La concentración y la calidad del ARN se determinaron por espectrofotometría en un NanoDrop ND-1000 (Thermo Fischer Scientific, Madrid, España) a 260-230nm. Los niveles relativos de cada ARN específico se determinaron mediante la técnica de transcriptasa reversa (RT) asociada a PCR en tiempo real (qPCR)22. A partir de 1μg de ARN total se obtuvo el ADN complementario que se amplificó mediante la reacción en cadena de la polimerasa usando 5-10ng de ADNc, SYBR green PCR Master Mix, primers específicos (detallados en la tabla 1) y el StepOne-Plus sequence detection system, todo ello de Applied Biosystems (Foster City, CA, EUA). Se utilizó tbp (TATA-box-binding protein) como control interno. Los resultados se calcularon usando el método 2-ΔCt.
Oligonucleótidos específicos para qPCR
| Forward primer (5’-3’) | Reverse primer (5’-3’) | |
|---|---|---|
| Aco | GTGAGGCGCCAGTCTGAAA | ACTGCTGGGTTTGAAAATCCA |
| Bip | TGATAATCAGCCCACCGTAACA | CAAATGTACCCAGAAGGTGATTGTC |
| Cat | CGCCTGTGTGAGAACATTGC | TAGTCAGGGTGGACGTCAGT |
| Chop | CCAGCAGAGGTCACAAGCAC | CGCACTGACCACTCTGTTTC |
| F4/80 | CAGATGGGGGATGACCACAC | AACTCTCCGTGGTGTCAGTG |
| Gadd34 | CTGGTCCAGGCAGACCATC | CTGTTTTGCTCACACCCAAGG |
| Gpx1 | TCGGTTTCCCGTGCAATC | TGAGGGAATTCAGAATCTCTTCATT |
| L-cpt-1 | CAGTGTGCCAGCCACAATTC | TAGGCTTCGTCATCCAGCAA |
| Pparα | TGGAGTCCACGCATGTGAAG | CGCCAGCTTTAGCCGAATAG |
| Sods2 | CGTCACCGAGGAGAAGTACCA | AGGCTGAAGAGCAACCTGAGTT |
| Tnfα | ATCCGAGATGTGGAACTGGC | CGATCACCCCGAAGTTCAGTA |
| Xbp1s | GAGTCCGCAGCAGGTG | CGTCAGAATCCATGGGAA |
| Xbp1 u | TCCGCAGCACTCAGACTACGT | ATGCCCAAAAGGATATCAGACTC |
| Tbp | TGGGATTGTACCACAGCTCCA | CTCATGATGACTGCAGCAAACC |
Los extractos de proteína total y proteína nuclear de hígado se obtuvieron según el método descrito previamente23. La cuantificación de la concentración proteica de cada fracción se realizó mediante el método de Bradford24.
30μg de proteína se sometieron a electroforesis en gel de poliacrilamida-SDS al 10%. Tras transferir las proteínas a membranas de PVDF (Immobilon-P polyvinylidene diflouride; Millipore Iberica, Bedford, MA, EUA) se bloquearon durante 1h a temperatura ambiente en solución de TBS-0.1% Tween-20 (TBS-Tween) con un 5% de leche en polvo desnatada. Las membranas se incubaron toda la noche a 4°C con el anticuerpo primario (PPARα de Abcam, dilución 1:500; p-IRE-1, IRE, p-PERK y PERK de Cell Signaling, dilución 1:1000 en TBS-Tween y un 5% de BSA) y 1h a temperatura ambiente con el anticuerpo secundario (antirabbit, dilución 1:3000). La detección se realizó utilizando el reactivo ECL chemiluminescence kit HRP (Thermo Fisher Scientific) y un ChemidocTM XRS de Bio-Rad (Hercules, CA, EUA). Como control de carga se incubaron las membranas con la proteína TATA-box-binding protein (TBP) (Abcam, Cambridge, UK, dilución 1:2500) o β-actina (Sigma-Aldrich, San Luis, MO, EUA, dilución 1:3000). El tamaño de las proteínas se comprobó utilizando marcadores de peso molecular de Bio-Rad. El software usado para la cuantificación relativa de las proteínas fue el ImageLab (BioRad).
Ensayo de binding o actividad de unión de PPARαPara la determinación de la actividad de unión del PPARα al DNA en muestras de extracto nuclear de hígado de rata, se utilizó el kit ab133107, PPAR alpha Transcription Factor assay kit de Abcam (Cambridge, UK). La técnica es un ELISA donde, en cada pozo de una placa de 96, se encuentra una secuencia de ADN específica que contiene el elemento de respuesta a PPAR (PPRE). Los PPAR presentes en los extractos nucleares (extractos obtenidos mediante el kit de Abcam, ab113474) se unen específicamente al PPRE y se cuantifican por colorimetría a 450nm. Se agregó al experimento un control positivo, un competidor de unión y blancos para confirmar que los procedimientos eran correctos. Los resultados se expresaron como % de unión del PPARα al ADN o PPRE.
EstadísticaLos resultados se expresan como la media±desviación estándar. Las muestras de plasma se estudiaron por duplicado. Las diferencias estadísticas se valoraron mediante un análisis de varianza (ANOVA) para muestras no apareadas y con análisis de Sidák post hoc, utilizando el programa informático GraphPad Prism (GraphPad Software V8.3). Cuando las desviaciones entre grupos no fueron homogéneas, se aplicó un ANOVA no paramétrico con postest de Kruskal-Wallis. Se consideró significación estadística un valor de p<0,05.
Resultados y discusiónEn las últimas décadas se ha observado un aumento de la prevalencia de NAFLD, que va acompañada de una mayor incidencia de patologías del metabolismo energético como son la diabetes y el síndrome metabólico1,25. La NAFLD es un espectro de trastornos que en sus fases iniciales se caracteriza por la hipertrigliceridemia y la acumulación de triglicéridos en el hígado (esteatosis hepática)2. Hasta el momento, y a pesar de la proporción casi epidémica que están alcanzando dichas alteraciones, no existe ningún fármaco aprobado para su tratamiento, lo que constituye claramente una necesidad sin cubrir en el campo médico.
Con el objetivo de buscar nuevas dianas terapéuticas para el tratamiento y prevención de la NAFLD y, a partir de estudios previos realizados por nuestro grupo de investigación19,22,26, diseñamos, para el presente trabajo, un modelo experimental de intervención dietética y farmacológica, como es la rata Sprague-Dawley hembra alimentada durante 3 meses con una dieta rica en grasa saturada y fructosa líquida al 10% (p/v), con el fin de inducir esteatosis hepática e hipertrigliceridemia. Se administró ácido bempedoico (ABem), por vía oral durante el último mes, para estudiar si estos efectos podían ser revertidos por el fármaco y elucidar otros mecanismos moleculares implicados, además de la inhibición de la ATP-citrato liasa (ACL) que es el mecanismo de acción propio del ácido bempedoico.
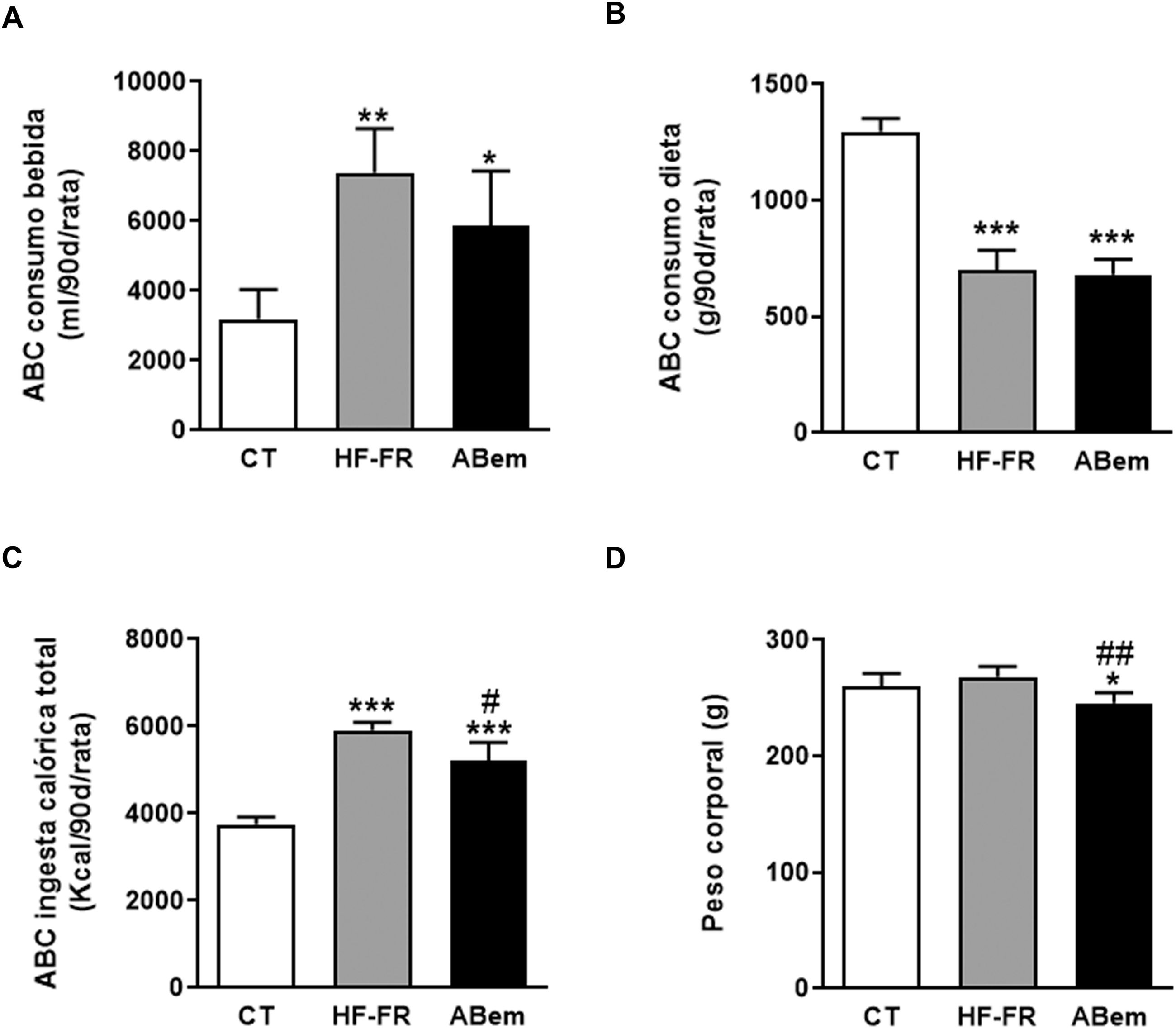
Como se muestra en la figura 1, el área bajo la curva del consumo de bebida demostró que los dos grupos de animales suplementados con un 10% (p/v) de fructosa, HF-FR y ABem, bebieron 2,3 y 1,8 veces más que los animales del grupo control, respectivamente (fig. 1A). Este incremento se acompañó de una menor ingesta de dieta sólida, que resultó ser estadísticamente significativa en ambos grupos (fig. 1B). Los cambios observados en el consumo de dieta y fructosa se tradujeron en un incremento en la ingesta calórica total que recibieron los animales HF-FR, siendo del 60% en el grupo HF-FR y del 38% en el grupo ABem, con respeto al grupo CT (fig. 1C). No obstante, la dieta HF-FR no modificó el peso corporal de los animales a lo largo del estudio, en comparación con el grupo CT (fig. 1D). En artículos previos habíamos descrito que ratones alimentados con una dieta western suplementada con fructosa líquida presentaban un incremento del peso corporal debido, principalmente, a un aumento en la ingesta calórica total22. Sin embargo, en el modelo actual no hemos observado este efecto. Este hecho puede deberse a la composición de la dieta HF que, a diferencia de la dieta western, no presenta colesterol y contiene una menor proporción de ácido palmítico, compensada con la inclusión de ácido esteárico (ácido graso saturado que se metaboliza a ácido oleico27). Esta peculiar composición, tal y como hemos descrito recientemente28, podría explicar que no aumentara el peso corporal de los animales a pesar de recibir alrededor de 1,6 veces más calorías que las ratas control.
 Área bajo la curva (ABC) del consumo de bebida por rata durante 90 días, expresado en ml (ml/90d/rata). (B) Área bajo la curva (ABC) del consumo de dieta sólida por rata durante 90 días, expresado en g (g/90d/rata). (C) Área bajo la curva (ABC) de la ingesta calórica total por rata durante 90 días, expresada en kcal (kcal/90d/rata). (D) Peso corporal de los animales a final de tratamiento expresado en g. Resultados expresados como media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. *p<0,05, **p<0,01, ***p<0,001 vs. grupo CT. #p<0,05, ##p<0,01 vs. grupo HF-FR.")
Consumo de bebida, comida, ingesta calórica total y peso de los animales. (A) Área bajo la curva (ABC) del consumo de bebida por rata durante 90 días, expresado en ml (ml/90d/rata). (B) Área bajo la curva (ABC) del consumo de dieta sólida por rata durante 90 días, expresado en g (g/90d/rata). (C) Área bajo la curva (ABC) de la ingesta calórica total por rata durante 90 días, expresada en kcal (kcal/90d/rata). (D) Peso corporal de los animales a final de tratamiento expresado en g. Resultados expresados como media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. *p<0,05, **p<0,01, ***p<0,001 vs. grupo CT. #p<0,05, ##p<0,01 vs. grupo HF-FR.
El tratamiento con ácido bempedoico no modificó el área bajo la curva del consumo de bebida ni de dieta, pero redujo la ingesta calórica total en un 12%, si lo comparamos con el grupo HF-FR (fig. 1). Así mismo, el fármaco redujo, de forma estadísticamente significativa, el peso corporal de los animales ABem en un 9% y un 10% vs. los grupos CT y HF-FR, respectivamente (fig. 1D). La reducción del peso corporal observada en el grupo ABem podría ser un efecto propio del fármaco no relacionado con la menor ingesta calórica de estos animales, ya que el grupo ABem mostró niveles de ingesta calórica total 1,38 veces los niveles del grupo control y una reducción del peso corporal del 9% vs. CT. Nuestros resultados están en consonancia con otros estudios publicados que demuestran que la administración de ABem a la dosis de 30mg/kg por día durante 3 semanas a roedores alimentados con una dieta rica en grasa y colesterol ven reducido su peso corporal respecto a los animales alimentados con la dieta sola14.
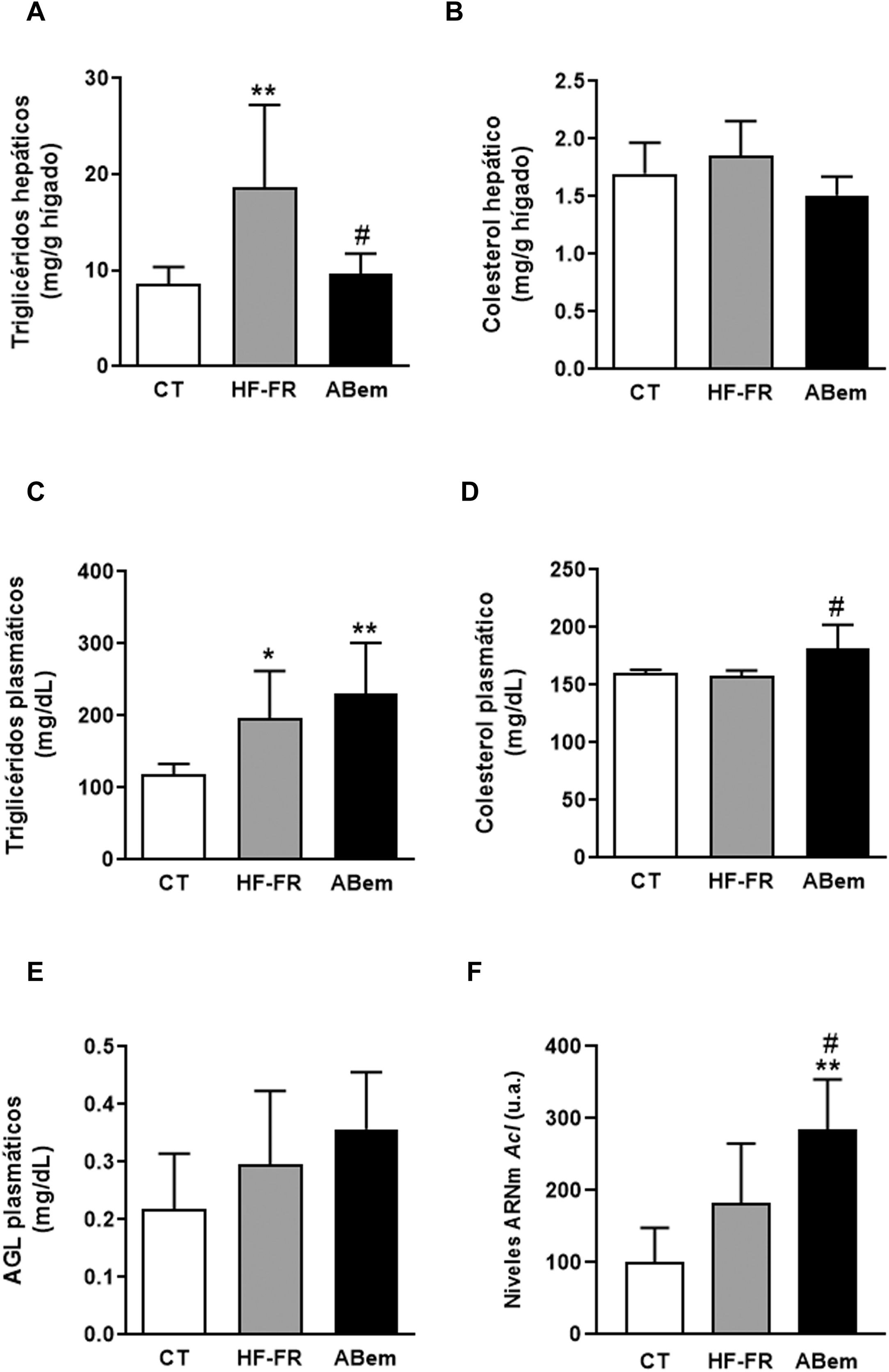
En nuestro estudio, la administración de dieta grasa y fructosa líquida durante 3 meses indujo la aparición de esteatosis hepática. El contenido de triglicéridos hepáticos se incrementó en el grupo HF-FR, siendo este incremento de 2,2 veces el de los animales control (fig. 2A). Así mismo, los análisis histopatológicos de secciones de hígado, realizados con tinción de hematoxilina y eosina, mostraron un mayor número de vesículas correspondientes a la acumulación de triglicéridos (fig. 3A). Por el contrario, no se observaron diferencias en los niveles de colesterol intrahepático en HF-FR con respecto al grupo CT (fig. 2B). En plasma, solo los niveles de triglicéridos se vieron fuertemente incrementados en un 66% en el grupo HF-FR vs. el grupo CT (fig. 2C), mientras que los niveles de colesterol y AGL no se modificaron con la dieta (fig. 2D y E).
 Contenido de triglicéridos hepáticos expresados como mg de triglicéridos/gramo de hígado. (B) Contenido de colesterol hepático expresado como mg de colesterol/gramo de hígado. (C) Niveles de triglicéridos plasmáticos. (D) Niveles de colesterol plasmático. (E) Niveles de ácidos grasos libres plasmáticos (AGL), expresados como mg de lípido/dl de plasma. (F) niveles de ARNm del gen Acl expresados en unidades arbitrarias (u.a.). Resultados expresados como media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. *p<0,05, **p<0,01 vs. grupo CT. #p<0,05 vs. grupo HF-FR.")
Lípidos plasmáticos y hepáticos. (A) Contenido de triglicéridos hepáticos expresados como mg de triglicéridos/gramo de hígado. (B) Contenido de colesterol hepático expresado como mg de colesterol/gramo de hígado. (C) Niveles de triglicéridos plasmáticos. (D) Niveles de colesterol plasmático. (E) Niveles de ácidos grasos libres plasmáticos (AGL), expresados como mg de lípido/dl de plasma. (F) niveles de ARNm del gen Acl expresados en unidades arbitrarias (u.a.). Resultados expresados como media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. *p<0,05, **p<0,01 vs. grupo CT. #p<0,05 vs. grupo HF-FR.
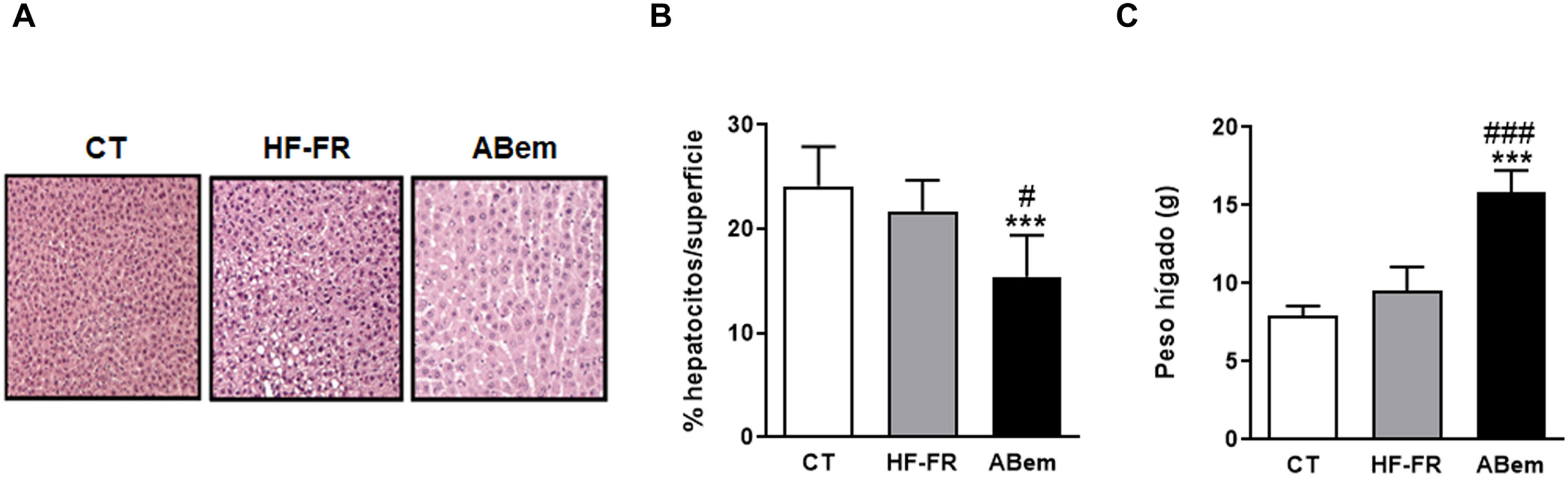
 Imagen representativa de la tinción por hematoxilina y eosina de cortes de hígado para los tres grupos de estudio. (B) Porcentaje (%) de núcleos de hepatocitos por superficie cuantificada para la valoración de la hipertrofia hepática. (C) Peso del hígado a final de tratamiento expresado en g. Los resultados son la media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. ***p<0,001 vs. grupo CT. #p<0,05, ###p<0,001 vs. grupo HF-FR.")
Hipertrofia hepática y peso del hígado. (A) Imagen representativa de la tinción por hematoxilina y eosina de cortes de hígado para los tres grupos de estudio. (B) Porcentaje (%) de núcleos de hepatocitos por superficie cuantificada para la valoración de la hipertrofia hepática. (C) Peso del hígado a final de tratamiento expresado en g. Los resultados son la media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. ***p<0,001 vs. grupo CT. #p<0,05, ###p<0,001 vs. grupo HF-FR.
Diversos autores describen que el ABem es capaz de reducir los niveles de colesterol plasmático en pacientes con hipercolesterolemia sin modificar los niveles de triglicéridos, a diferencia de lo observado en modelos de roedores16,17. Sin embargo, en nuestro modelo experimental, el fármaco no modificó la hipertrigliceridemia inducida por la dieta (fig. 2C), e incrementó los niveles de colesterol un 15% con respecto al grupo HF-FR (fig. 2D). Nuestra hipótesis es que ABem podría estar induciendo un mayor transporte de lípidos del hígado al plasma a través de las VLDL, lo que justificaría el aumento de los niveles plasmáticos de colesterol y triglicéridos, aunque este no es significativo en el último caso.
El tratamiento con ABem revirtió la esteatosis hepática inducida por la dieta HF-FR, disminuyendo el contenido de triglicéridos en hígado un 51% vs. el grupo HF-FR, sin modificar los niveles intrahepáticos de colesterol (fig. 2A y B). El ácido bempedoico reduce la síntesis de colesterol y ácidos grasos en el hígado por un mecanismo que implica la inhibición de la ATP-citrato liasa (ACL)15,29. En nuestros animales, los niveles de ARNm de ACL se vieron fuertemente incrementados en el grupo ABem (1.56X vs. HF-FR y 2.85X vs. CT) (fig. 2F). Esta inducción en la expresión de ACL podría ser un mecanismo para compensar la inhibición de la actividad enzimática provocada por el fármaco. La inhibición de la ACL comporta una menor síntesis hepática de ácidos grasos y, por tanto, de triglicéridos. Ello estaría de acuerdo con la reducción en el contenido de triglicéridos hepáticos observado en el grupo ABem vs. HF-FR.
A pesar de la mejoría observada tras el tratamiento con ácido bempedoico sobre la esteatosis hepática, los hígados del grupo ABem mostraron una marcada hipertrofia de los hepatocitos (fig. 3B) reduciendo el % de núcleos celulares por área cuantificada en un 30% con respeto al grupo CT y un 36% respeto al grupo HF-FR. Paralelamente, el peso del hígado en el grupo ABem se vio fuertemente incrementado, siendo 1.7X el de los animales HF-FR (fig. 3C). Ambos efectos no pudieron ser causados por la acumulación de lípidos hepáticos ya que, como ya se ha comentado, los niveles de triglicéridos estaban reducidos en los hígados del grupo ABem y los niveles de colesterol no se modificaron (fig. 2 A y B).
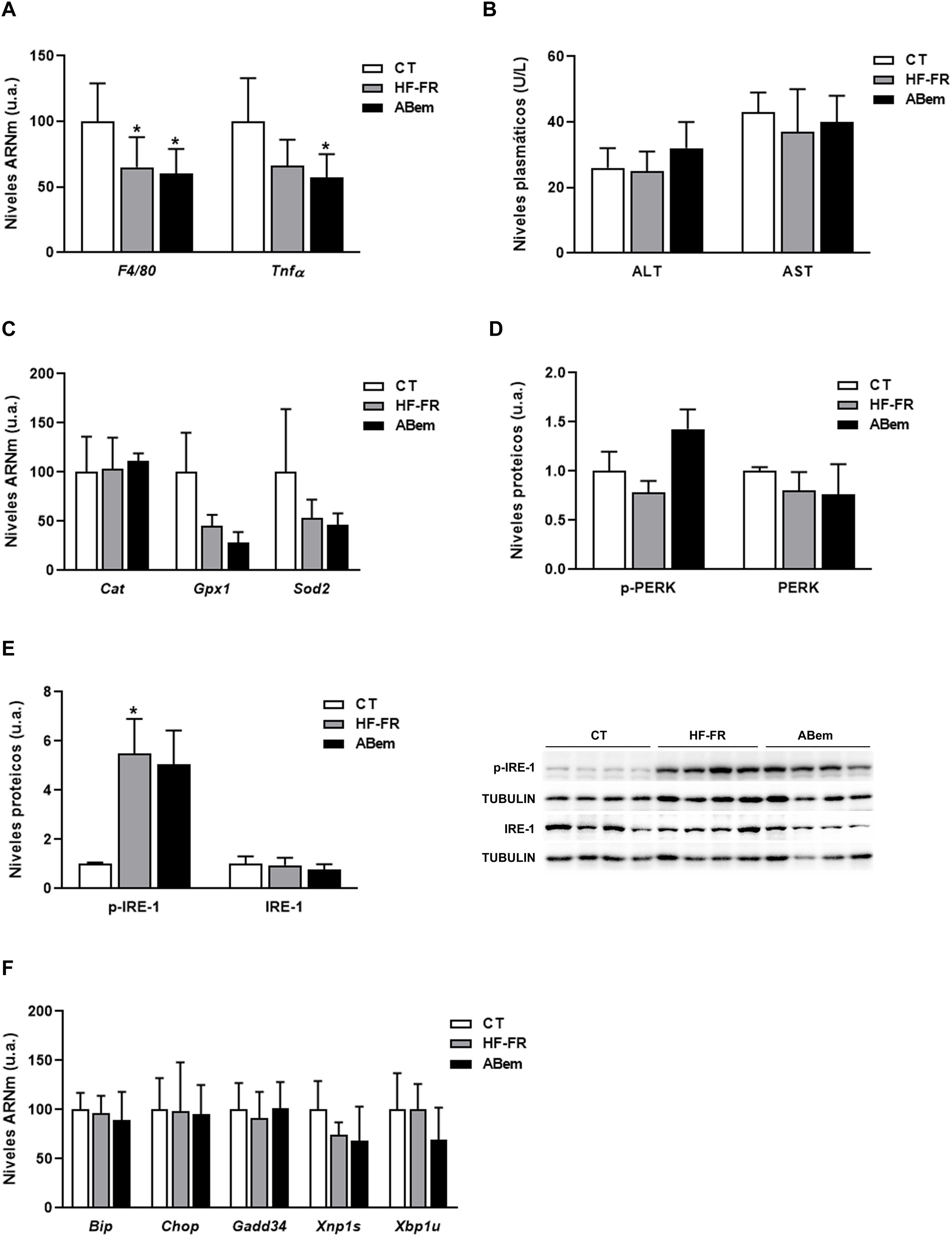
La inflamación hepática, el aumento en los marcadores de estrés oxidativo y el estrés de retículo endoplasmático son complicaciones propias de la NAFLD30,31. Por ello, analizamos si la hipertrofia hepática y el incremento de peso del hígado observado en los animales ABem eran debidos a una alteración en alguna de estas tres vías. Tal y como se muestra en la figura 4A, los niveles de ARNm de biomarcadores de inflamación como Tnf-α y F4/80 se vieron reducidos en los dos grupos de animales alimentados con dieta HF-FR (35% en HF-FR y 40% en ABem vs. CT). La administración del fármaco no produjo modificaciones en la expresión de ninguno de estos genes cuando se comparó con el grupo HF-FR. Los niveles plasmáticos de AST y ALT, marcadores bioquímicos de lesión hepática e inflamación, tampoco mostraron diferencias entre los grupos de estudio (fig. 4B). Así mismo, los niveles de expresión de Cat, Gpx1 y Sods2, marcadores de estrés oxidativo, no se modificaron en ninguno de los dos grupos, HF-FR y ABem (fig. 4C). Al analizar las vías de señalización de estrés de retículo, IRE-1 y PERK, observamos que únicamente los niveles proteicos de p-IRE-1 se incrementaron en muestras de hígado de animales HF-FR (fig. 4D), mientras que los niveles de p-PERK no se modificaron (fig. 4E). A pesar de la activación por fosforilación de IRE-1, no se produjo ningún cambio significativo en la expresión génica de los marcadores Bip, Chop, Gadd34, Xbp1s y Xbp1u, propios de estas dos vías, en los hígados de ratas HF-FR vs. CT (fig. 4F). La administración de ABem no modificó de forma estadísticamente significativa los niveles proteicos de p-IRE-1 ni ninguno de los genes estudiados, si los comparamos con el grupo HF-FR (fig. 4D y F). Estos resultados nos permiten confirmar que nuestro modelo dietético induce hígado graso, propio de las primeras etapas de NALFD, pero evita el efecto proinflamatorio característico de las dietas ricas en grasa32, ya que no induce inflamación, estrés oxidativo ni estrés de retículo endoplasmático. El tratamiento con ABem tampoco modifica ninguno de estos parámetros cuando lo comparamos con los animales CT.
 Niveles de ARNm de genes marcadores de inflamación expresados en unidades arbitrarias (u.a.). (B) Niveles plasmáticos de ALT y AST expresados en unidades/litro de plasma (U/L). (C) Niveles de ARNm de genes marcadores de estrés oxidativo expresados en unidades arbitrarias (u.a.). (D) Niveles de proteína PERK (fosforilada y total) expresados en unidades arbitrarias (u.a.) (E) Niveles proteicos de IRE-1 (fosforilado y total) expresados en unidades arbitrarias (u.a.). A la derecha del gráfico se muestra una imagen representativa del experimento de western blot realizado para p-IRE1 e IRE-1 para n=3 muestras por grupo de estudio. (F) Niveles de ARNm de genes clave de las diferentes vías de estrés de retículo, expresados en unidades arbitrarias (u.a.). Los resultados son la media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. *p<0,05 vs. grupo CT.")
Marcadores de inflamación, estrés oxidativo y estrés de retículo. (A) Niveles de ARNm de genes marcadores de inflamación expresados en unidades arbitrarias (u.a.). (B) Niveles plasmáticos de ALT y AST expresados en unidades/litro de plasma (U/L). (C) Niveles de ARNm de genes marcadores de estrés oxidativo expresados en unidades arbitrarias (u.a.). (D) Niveles de proteína PERK (fosforilada y total) expresados en unidades arbitrarias (u.a.) (E) Niveles proteicos de IRE-1 (fosforilado y total) expresados en unidades arbitrarias (u.a.). A la derecha del gráfico se muestra una imagen representativa del experimento de western blot realizado para p-IRE1 e IRE-1 para n=3 muestras por grupo de estudio. (F) Niveles de ARNm de genes clave de las diferentes vías de estrés de retículo, expresados en unidades arbitrarias (u.a.). Los resultados son la media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. *p<0,05 vs. grupo CT.
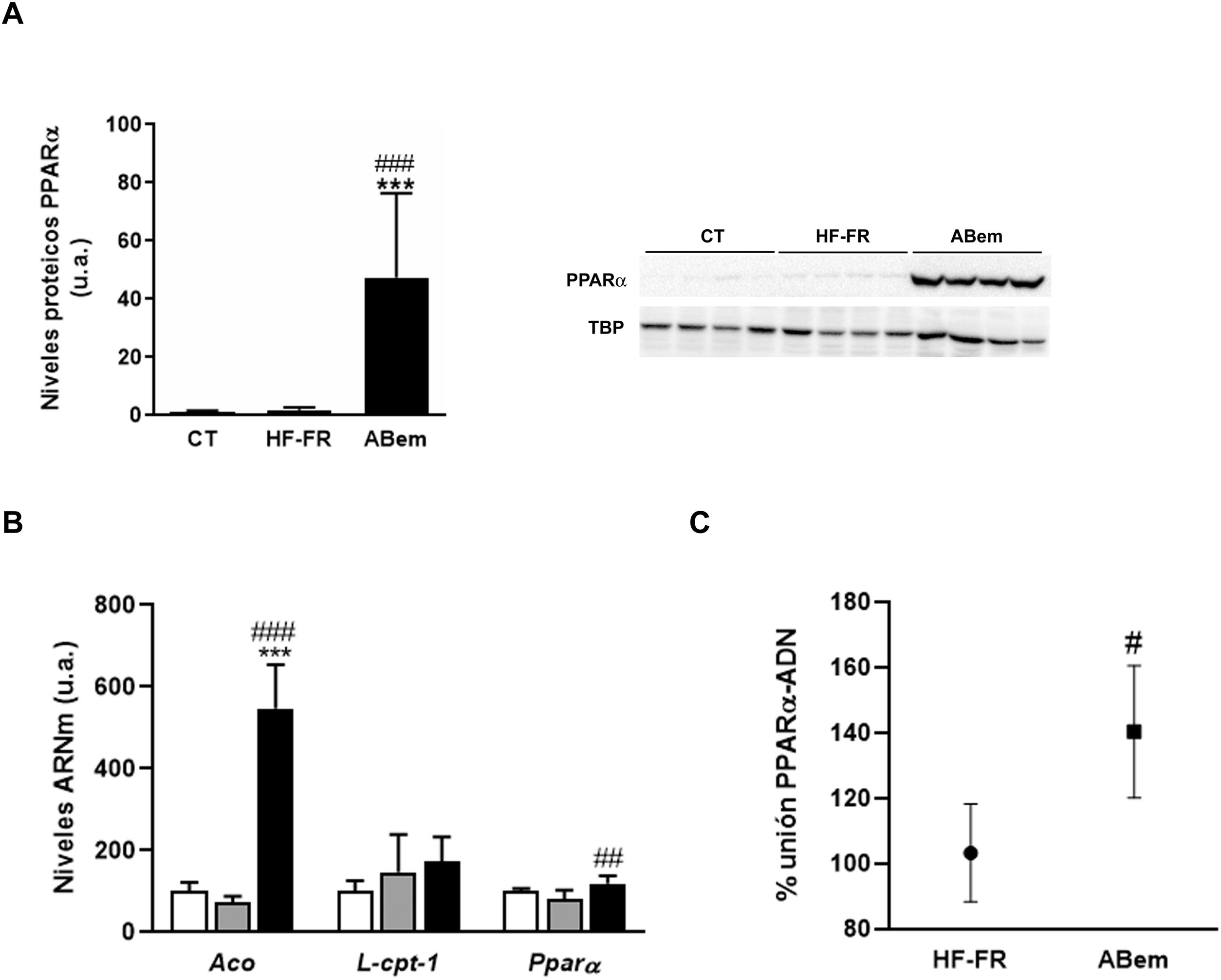
El fenómeno de proliferación peroxisómica es otra causa de hipertrofia hepatocelular que cursa con un aumento del tamaño y número de peroxisomas y con un incremento en la actividad de β-oxidación de los ácidos grasos y de la expresión de genes relacionados con esta vía y regulados por PPARα33,34. Los fibratos, fármacos hipolipemiantes agonistas del receptor nuclear PPARα, son conocidos inductores de este fenómeno de proliferación peroxisómica en roedores35. Estudios realizados por Samsoondar et al.17, en un modelo murino, sugirieron que el ABem podría actuar de manera similar a un proliferador peroxisómico; el aumento observado en el peso del hígado de los animales tratados con ABem se correlacionó con un ligero efecto de proliferación peroxisómica, un incremento en la β-oxidación de los ácidos grasos y un aumento en la expresión de Aco (acil-CoA oxidasa, enzima velocidad limitante de la reacción de β-oxidación peroxisomal y diana de PPARα). Nuestros resultados concuerdan con dicha hipótesis. El consumo de dieta HF-FR no modificó la expresión proteica de PPARα ni la de sus genes diana (fig. 5A y B). Sin embargo, el tratamiento con ABem incrementó la expresión de PPARα, tanto los niveles de proteína hepáticos (25X vs. HF-FR) (fig. 5A) como los niveles de ARNm (1.46X vs. HF-FR). La activación de PPARα por parte del fármaco incrementó los niveles de ARNm de sus genes diana, siendo la expresión génica de Aco 7.4X en el grupo ABem vs. el grupo HF-FR (fig. 5B). Para poder elucidar si el ABem se comporta, en nuestro modelo, como un activador directo de PPARα, realizamos estudios de binding o de actividad de unión del PPARα a su elemento de respuesta (PPRE), en muestras hepáticas de animales HF-FR y ABem, utilizando el kit comercial PPAR alpha Transcription Factor assay kit de Abcam. Los animales tratados con ABem presentaron un 40% más de capacidad de unión de PPARα a su PPRE que los animales del grupo HF-FR, tal y como se muestra en la figura 5C. Todos estos resultados en su conjunto nos sugieren que el ácido bempedoico se comportaría como un activador directo del receptor PPARα. La activación PPARα contribuiría de forma significativa al efecto de ABem sobre la esteatosis hepática inducida por la dieta, y al mismo tiempo, podría ser la responsable de la hipertrofia de los hepatocitos y el incremento del peso del hígado observado en los animales tratados.
 Niveles de proteína de PPARα expresados en unidades arbitrarias (u.a.). Se muestra una imagen representativa del ensayo de western blot realizado para n=4 muestras por grupo de estudio. Los resultados se normalizaron usando la proteína TATA-box-binding protein (TBP) como control. (B) Niveles de ARNm de genes diana de PPARα expresados en unidades arbitrarias (u.a.). (C) Actividad de unión de PPARα a PPRE expresada como porcentaje (%) de unión en los grupos HF-FR y ABem. Resultados expresados como media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. ***p<0,001 vs. grupo CT. #p<0,05, ##p<0,01, ###p<0,001 vs. grupo HF-FR.")
Determinación de la expresión y actividad PPARα (A) Niveles de proteína de PPARα expresados en unidades arbitrarias (u.a.). Se muestra una imagen representativa del ensayo de western blot realizado para n=4 muestras por grupo de estudio. Los resultados se normalizaron usando la proteína TATA-box-binding protein (TBP) como control. (B) Niveles de ARNm de genes diana de PPARα expresados en unidades arbitrarias (u.a.). (C) Actividad de unión de PPARα a PPRE expresada como porcentaje (%) de unión en los grupos HF-FR y ABem. Resultados expresados como media±DE para n=8 animales/grupo. ABem: grupo alimentado con HF-FR y tratado con ácido bempedoico; CT: grupo control; HF-FR: grupo alimentado con dieta rica en grasa suplementada con fructosa en el agua de bebida. ***p<0,001 vs. grupo CT. #p<0,05, ##p<0,01, ###p<0,001 vs. grupo HF-FR.
Nuestros resultados nos permiten concluir que la administración a ratas Sprague-Dawley hembra de una dieta rica en ácidos grasos y desprovista de colesterol, suplementada con un 10% (p/v) de fructosa en el agua de bebida durante 3 meses, induce esteatosis hepática e hipertrigliceridemia, sin alterar los marcadores proinflamatorios, inducir estrés oxidativo o alterar las vías de estrés de retículo endoplasmático, lo que indica que nos encontramos en las fases iniciales de la NAFLD. El ácido bempedoico revierte la esteatosis hepática por un mecanismo que implicaría, al menos en parte, la activación directa del receptor nuclear PPARα. Este mecanismo difiere, pero posiblemente no excluye, la inhibición de la ACL, mecanismo principal de acción del ácido bempedoico.
FinanciaciónEl presente trabajo ha sido financiado por la Sociedad Española de Arteriosclerosis mediante la concesión de una beca FEA/SEA 2020 Investigación Básica en Arteriosclerosis y por un proyecto del Ministerio de Ciencia e Innovación, referencia: SAF2017-82369.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.