




La aterosclerosis y sus consecuencias clínicas (enfermedad cardíaca coronaria y accidente cerebrovascular) son las principales causas de muerte no sólo en los países desarrollados, sino también en países con ingresos medios o bajos, y las previsiones para el año 2030 parecen seguir la misma tendencia1. En la actualidad, las principales aproximaciones terapéuticas destinadas a reducir el riesgo cardiovascular se basan en la corrección de las dislipemias mediante inhibidores de la 3-hidroxi-3-metil glutaril coenzima A reductasa (estatinas) y agonistas de los receptores activados por proliferadores peroxisómicos (PPAR) a (fibratos). Sin embargo, los avances en la comprensión de las vías moleculares y de los procesos celulares implicados en la aterogénesis impulsan al desarrollo de nuevas estrategias que puedan actuar de forma independiente o adicional a la reducción de los lípidos plasmáticos. Una de estas estrategias se basa en el control directo de los procesos que tienen lugar en las propias células que formarán la placa de ateroma, entre las cuales destacan los monocitos y los macrófagos. Estas células desempeñan un papel clave en todas las fases del desarrollo de la aterosclerosis, desde sus inicios hasta la rotura de la placa de ateroma, por lo cual constituyen dianas terapéuticas idóneas para una acción antiaterosclerótica. En esta revisión se aborda la contribución de monocitos y macrófagos a cada una de las etapas de la formación de la placa de ateroma, los mecanismos moleculares implicados y las potenciales dianas farmacológicas en cada caso.
Adhesión y migración de monocitos: fenómenos iniciales en la aterogénesis
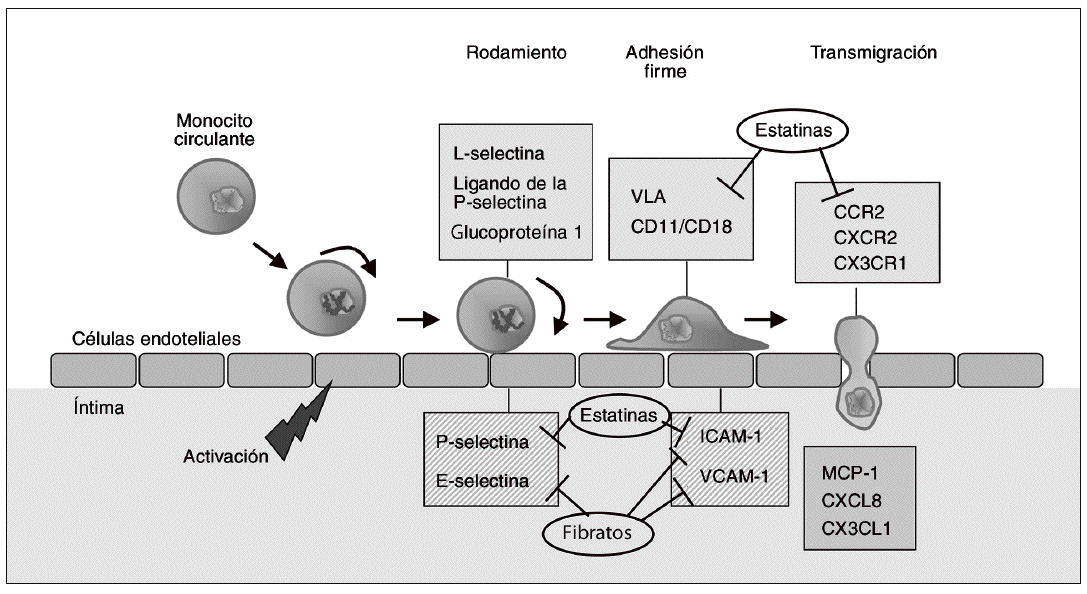
El reclutamiento de monocitos circulantes hacia la capa íntima arterial constituye uno de los fenómenos más tempranos en el desarrollo de la aterosclerosis. La disfunción y la activación de las células endoteliales, que resulta de un intento de adaptación a estímulos anormales (valores elevados de lipoproteínas de baja densidad [LDL], LDL modificadas, radicales libres, fuerzas hemodinámicas, concentraciones elevadas de homocisteína o combinaciones de estos factores)2, conduce a un incremento en la expresión de moléculas de adhesión celular (MA). Las MA median la adherencia y posterior transmigración de los leucocitos mononu-cleares (monocitos y células T) a través de la superficie endotelial, por lo que desempeñan un papel destacado en la génesis de la aterosclerosis3. Inicialmente, los monocitos se adhieren de forma lábil al endotelio vascular, proceso denominado "rodamiento" o "adhesión rodante", que es mediado por las selectinas expresadas por las células endoteliales (E- y P-selectinas) y por los monocitos (L-selectina). El rodamiento causa la disminución de la velocidad de los monocitos y permite la interacción entre las integrinas leucocitarias (very late-activation antigen [VLA], CD11a/CD18 [LFA-1] y CD11b/CD18 [Mac-1]), y las MA de la superfamilia de las inmunoglobulinas expresadas por las células endoteliales4. Esta familia incluye la molécula de adhesión intercelular de tipo 1 (ICAM-1) y la molécula de adhesión celular vascular-1 (VCAM-1). Tanto en modelos animales de aterosclerosis como en lesiones ateroscleróticas humanas, dichas MA se hallan sobreexpresadas en relación con el tejido vascular normal5. Además, las concentraciones plasmáticas de MA solubles son mucho más elevadas en situaciones de hipertensión, obesidad, dislipemia y resistencia a la insulina3, así como en pacientes con enfermedad coronaria cardíaca6, en comparación con controles sanos.
Dado que la adhesión de los monocitos es una de las etapas más precoces en el desarrollo de la aterosclerosis, la inhibición de la expresión de MA es una opción de potencial utilidad antiaterogénica. La figura 1 muestra algunas de las dianas farmacológicas en esta fase inicial de la formación de lesiones ateroscleróticas. La inhibición directa de la función de las MA ha atraído un interés considerable en los últimos años, no sólo en el caso de la aterosclerosis sino en otras enfermedades de componente inflamatorio como el asma, la enfermedad inflamatoria intestinal o la artritis reumatoide7,8. El bloqueo de selectinas mediante anticuerpos selectivos dio buenos resultados en los estudios preclínicos iniciales, pero los resultados obtenidos en la fase clínica han sido inconsistentes. De forma similar, los ensayos clínicos con anticuerpos contra CD18 (rovelizumab o erlizumab) o contra ICAM-1 (enlimomab) no han demostrado efectos beneficiosos en la isquemia-reperfusión después de infarto de miocardio o accidente cerebrovascular8. A pesar de estos resultados decepcionantes, lo que sí parece evidente es que los beneficios cardiovasculares de diversos fármacos utilizados actualmente en clínica derivan, al menos en parte, de su capacidad para modificar la expresión de MA, y por consiguiente inhibir la acumulación de monocitos en la íntima arterial. Así, las estatinas reducen la expresión de integrinas como VLA, CD11a/b o CD189,10, así como la expresión de ICAM-111,12 y P-selectina en las células endoteliales13. Estos efectos parecen ser independientes de la reducción de lípidos plasmáticos, y se han relacionado con la reducción de la síntesis de intermediarios de tipo isoprenoide derivados del mevalonato que se produce como consecuencia de la inhibición de la enzima hidroxi-metil glutaril coenzima A (HMG-CoA) reductasa; ello comporta la inhibición de la isoprenilación de proteínas G pequeñas, como Rho, y por tanto de su activación14. Por otra parte, las estatinas pueden reducir la adhesión de linfocitos al endotelio a través de su unión a zonas reguladoras del antígeno LFA-1, de forma que estabilizan este receptor en su forma inactiva15,16. Este efecto no sólo es independiente de la reducción de lípidos, sino que tampoco se relaciona con la inhibición de la HMG-CoA reductasa y la isoprenilación de proteínas17. Los agonistas PPARa, como los fibratos, también reducen la expresión de VCAM-1 e ICAM-1 inducida por citocinas proinflamatorias en el endotelio vascular, y reducen la concentración de ICAM-1 y E-selectina circulantes18. Dichos efectos están mediados, al menos en parte, por la interferencia con el factor proinflamatorio nuclear kB (NFkB). También los agonistas PPARg, como la rosiglitazona, reducen la expresión de moléculas de adhesión en estudios in vitro y en modelos animales de isquemia de miocardio19,20. Un estudio reciente en un modelo animal de isquemia-reperfusión sugiere que los efectos de la rosiglitazona ocurren básicamente durante la transición hacia el proceso de adhesión firme, ya que el fármaco reduce de forma marcada el ARNm y la proteína de VCAM-121. Por otra parte, los bloqueadores de canales de calcio de tipo L, como el amlodipino, ampliamente utilizados en el tratamiento de la hipertensión y la enfermedad cardíaca coronaria, han demostrado ser capaces de inhibir la progresión de la aterosclerosis en modelos animales22. En ratones que no expresan apolipoproteína E (apo E/) alimentados con dieta rica en colesterol, el tratamiento concomitante con amlodipino impide el incremento en la expresión de moléculas de adhesión como ICAM-1 y VCAM-1, lo que podría explicar en parte su efecto ateroprotector23.
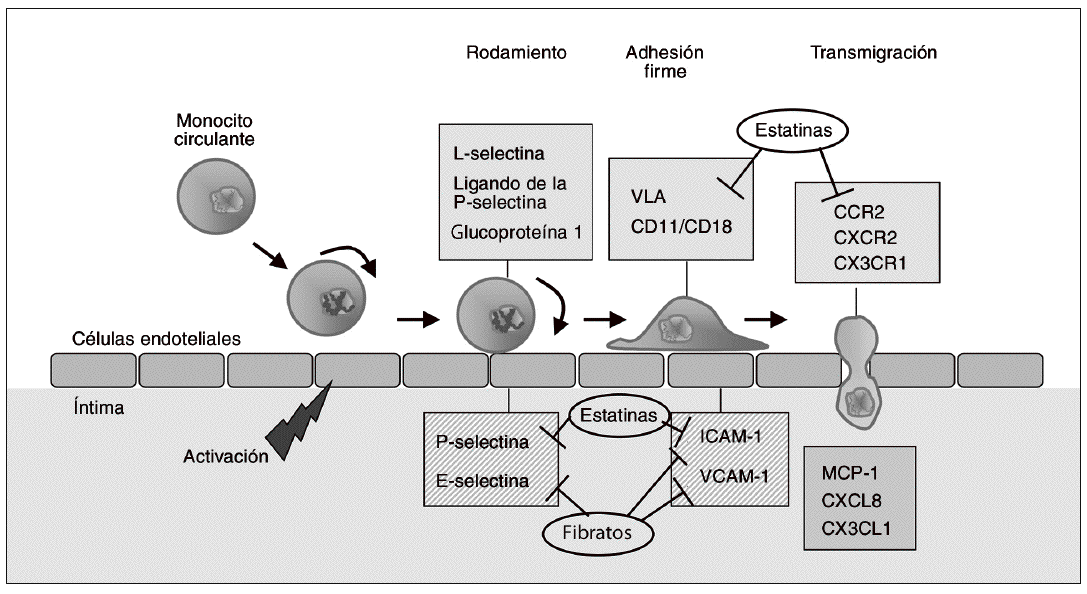
Figura 1. Dianas terapéuticas en fases iniciales del desarrollo de la aterosclerosis. La activación de células endoteliales conduce a un incremento de la expresión de moléculas de adhesión en estas mismas células (E- y P-selectinas) y de sus ligandos en la superficie de los monocitos (L-selectina y ligando de la P-selectina glucoproteína-1). La interacción entre selectinas y sus ligandos hace que los monocitos se deslicen rodando lentamente por la superficie del endotelio (rodamiento o adhesión rodante). Esto facilita la interacción entre las integrinas del monocito (very late-activation antigen [VLA] y heterodímeros CD11/CD18), y las proteínas de la familia de las inmunoglobulinas expresadas en las células endoteliales, como la molécula de adhesión intercelular 1 (ICAM-1) y la molécula de adhesión celular vascular 1 (VCAM-1), que median la adhesión firme. Posteriormente, los monocitos migran hacia la íntima en un proceso estimulado por quimiocinas como la proteína quimioatrayente de monocitos (MCP-1), CXCL8 y CX3CL1, que interactúan con sus respectivos receptores CCR2, CXCR2 y CX3CR1. La inhibición de la expresión de moléculas de adhesión quimiocinas o sus receptores resulta en una menor infiltración de monocitos que, teniendo en cuenta el papel de estas células en la aterosclerosis, puede reducir el desarrollo de lesiones. La figura muestra los puntos de actuación de fármacos de uso clínico (estatinas y fibratos) sobre estas dianas antiateroscleróticas tempranas.
Una vez los monocitos se han adherido firmemente al endotelio, migran hacia la íntima en un proceso estimulado por quimiocinas sintetizadas por células de la pared arterial, como células endoteliales y células musculares lisas, así como por los propios macrófagos. Las quimiocinas son polipéptidos de pequeño tamaño que actúan a través de la interacción con receptores expresados en la superficie de los monocitos. Una de las mejor caracterizadas es la proteína quimioatrayente de monocitos (MCP-1), también denominada CCL2, que interactúa con el correspondiente receptor monocítico CCR2 y desempeña un papel crítico en el reclutamiento de monocitos a través de la pared arterial4,24. Otras quimiocinas importantes en este proceso son CXCL8 y CX3CL1, que se unen a sus respectivos receptores CXCR2 y CX3CR125,26. Tanto las citocinas proinflamatorias como las LDL modificadas son capaces de incrementar la expresión de MCP-1 en las células endoteliales, facilitando la transmigración de los monocitos a la íntima arterial27. Por otra parte, la presencia de MCP-1 en lesiones ateroscleróticas humanas está bien documentada28. Así pues, MCP-1 constituye otra de las dianas terapéuticas "tempranas" cuya modulación puede reducir el desarrollo de la aterosclerosis (fig. 1). De hecho, diversos estudios han demostrado que tanto la terapia génica contra esta quimiocina como la deleción del receptor CCR2 limitan la progresión de la aterosclerosis en ratones apo E/29,30. En la actualidad, terapias destinadas a bloquear la interacción de CCR2 con sus ligandos están siendo evaluadas en el ámbito clínico8.
Por otra parte, algunos fármacos utilizados actualmente, como las estatinas, reducen la expresión de CCR2 en los monocitos, inhibiendo así la migración de estas células31-33. Este efecto se atribuye a la inhibición de la HMG-CoA reductasa en los monocitos circulantes, con la consiguiente reducción de los valores de geranilgeranil pirofosfato e inactivación de Rho A, lo que causa un incremento en la actividad PPARg. En este sentido, otros estudios han demostrado que los agonistas PPARg son capaces de reducir la expresión de CCR2 en monocitos THP-134,35, ya que reprimen la expresión del promotor de este gen36.
Determinados componentes lipídicos presentes en las LDL oxidadas reducen la expresión de CCR2 en monocitos por un mecanismo dependiente de PPARg; este efecto bloquea la migración reversa del monocito, promoviendo su retención en la íntima arterial35. Recientemente se ha propuesto que la activación PPARg por los lípidos oxidados promueve un cambio en la expresión de receptores (reducción de CCR2 e incremento de CX3CR1) que conduce al cese de la migración del monocito y activa los mecanismos de retención mediados por CX3CR1, de modo que se facilita la acumulación de monocitos/macrófagos en la pared del vaso. La expresión de CX3CL1 y de su receptor CX3CR1 es elevada en la enfermedad arterial coronaria y se reduce por el tratamiento con estatinas37. Todo ello sugiere que el sistema CX3CL1/CX3CR1 constituye otra diana terapéutica de potencial interés (fig. 1).
En síntesis, la modulación terapéutica de MA o de quimiocinas puede inhibir el reclutamiento de monocitos, una de las fases iniciales de la aterogénesis. Sin embargo, debe tenerse en cuenta que el movimiento de monocitos a través de la íntima arterial es un proceso dinámico que tiene lugar a lo largo del desarrollo de la lesión, de modo que la inhibición del reclutamiento puede también influir fases más tardías del mismo38.
Inflamación e inmunidad
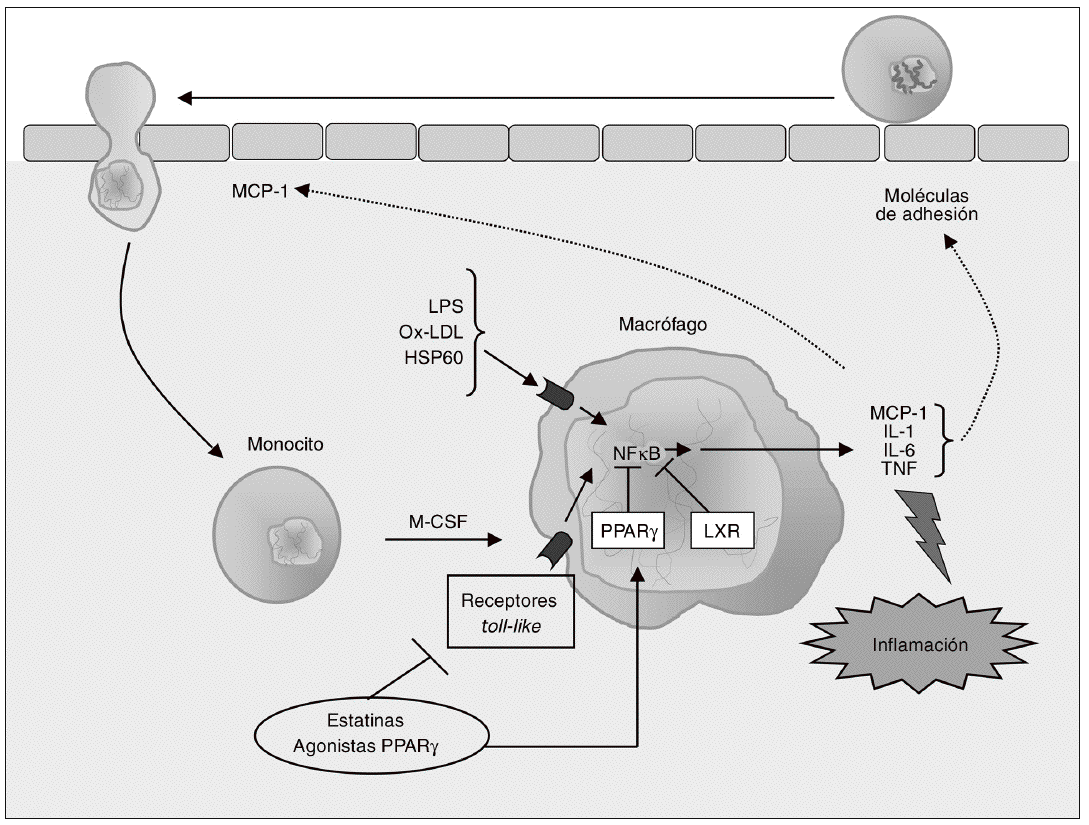
Los monocitos que han sido reclutados se diferencian a macrófagos bajo la influencia del factor estimulador de colonias de macrófagos (M-CSF), una citocina formada en la propia capa íntima inflamada24 (fig. 2). La importancia del M-CSF en la aterosclerosis se evidenció a partir de estudios realizados en ratones op/op, un modelo de deficiencia de M-CSF caracterizado por una carencia de macrófagos diferenciados en diversos órganos y tejidos. Cuando estos ratones se cruzan con ratones apo E/, la descendencia presenta escasas lesiones ateroscleróticas, lo que confirma que la diferenciación de monocito a macrófago es un paso necesario en el desarrollo de aterosclerosis39.
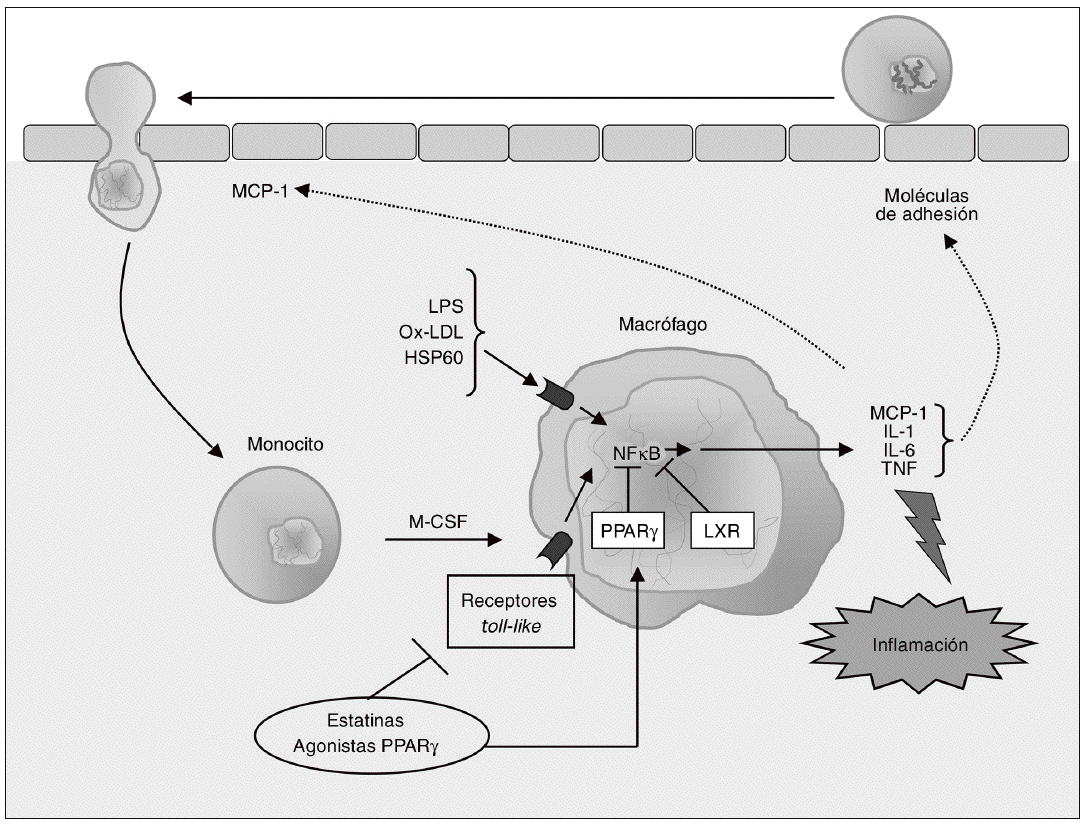
Figura 2. Modulación de la respuesta inflamatoria en el macrófago. Los monocitos que han sido reclutados se diferencian a macrófagos bajo la influencia del factor estimulante de colonias de macrófagos (M-CSF), proceso que se asocia con el incremento en la expresión de receptores toll-like (TLR), implicados en la respuesta inflamatoria e inmune. La activación de TLR en los macrófagos inicia una cascada de señalización mediada por el factor nuclear kB (NFkB) que conduce a la liberación de citocinas proinflamatorias (interleucina [IL]-1, IL-6 y factor de necrosis tumoral alfa [TNF-a]) y de quimiocinas como la MCP-1, que contribuyen al establecimiento de una respuesta inflamatoria local lesiva para el tejido. Las citocinas secretadas por el macrófago estimulan la expresión de moléculas de adhesión, de forma que se reclutan más monocitos, creándose un círculo vicioso que amplifica y perpetúa la reacción inflamatoria. Los receptores activados por proliferadores peroxisómicos (PPAR) g y los receptores hepáticos X (LXR) son reguladores negativos de la respuesta inflamatoria en el macrófago, por lo que constituyen dianas terapéuticas de gran interés para modular farmacológicamente la secreción de citocinas en estas células.
La adquisición del fenotipo de macrófago se asocia con un incremento en la expresión de receptores de estructuras microbianas (pattern-recognition receptors), incluyendo los receptores toll like (TLR) y los receptores scavenger (SR). Los TLR del macrófago desempeñan un importante papel en la respuesta inmune que caracteriza el proceso aterosclerótico, y activan la respuesta inflamatoria en estas células a través del sistema NFkB40 (fig. 2). Hasta el momento se han identificado como mínimo 10 TLR para los cuales se conocen los ligandos activadores41; entre ellos, el TLR4 es uno de los mejor caracterizados en relación con el desarrollo de la aterosclerosis.
TLR4 reconoce lipopolisacáridos bacterianos (LPS) y otros ligandos relevantes para la aterogénesis, como las LDL oxidadas y las proteínas de choque térmico (heat-shock proteins) como la Hsp60. El receptor TLR4 se expresa en diversos tipos celulares de la pared arterial, y se detecta en lesiones ateroscleróticas humanas, principalmente colocalizado con macrófagos42. Además, los pacientes con síndrome coronario agudo muestran una elevada expresión de TLR4 en la superficie de los monocitos43, y la presencia de un polimorfismo en el gen que codifica para TLR4 (Asp299Gly) se ha asociado con una menor incidencia de aterosclerosis44. Éstas y otras evidencias sugieren que TLR4 está implicado en distintas fases del desarrollo de la aterosclerosis. Por ejemplo, en fases iniciales la activación de TLR4 causa un aumento en la expresión de MCP-1 y otras quimiocinas, de modo que se estimula el reclutamiento de monocitos42. En consonancia con un papel proaterogénico para TLR4, el déficit genético de este receptor se asocia con una reducción en el tamaño y en el contenido lipídico de las lesiones ateroscleróticas en ratones apo E/ hipercolesterolémicos, sin que existan variaciones en las concentraciones de colesterol plasmático45. En cambio, la deficiencia selectiva de TLR4 en macrófagos no altera el desarrollo de aterosclerosis en ratones LDLR/40.
A pesar de estos resultados contradictorios, TLR4 puede considerarse una diana para la intervención terapéutica en la aterosclerosis. Sin embargo, debido al papel de este receptor en la respuesta inmune y en la defensa contra patógenos, la inhibición sistémica de TLR4 causaría graves complicaciones, como un aumento de susceptibilidad ante las infecciones bacterianas46. El tratamiento local con agentes farmacológicos, ARN de interferencia o terapia génica para inhibir la señalización mediada por TLR4 podrían ser aproximaciones más adecuadas. En este aspecto, puede destacarse que las estatinas reducen la expresión de TLR4 en la superficie de los monocitos in vitro e in vivo, inhibiendo así la expresión de citocinas proinflamatorias; estos efectos están mediados por la inhibición de la geranilgeranilación y farnesilación de proteínas47. Recientemente, Niessner et al48 demostraron que el pretratamiento con simvastatina en un modelo humano de endotoxemia reduce la expresión de TLR4 y TLR2, otro receptor toll like implicado en la aterosclerosis, en monocitos49. Por otra parte, se ha demostrado que también los agonistas PPARg reprimen genes diana de TLR en macrófagos en cultivo e in vivo a través de mecanismos de transrepresión específicos50.
Como ya se ha comentado anteriormente, la activación de los TLR en macrófagos inicia una cascada de señalización que conduce a la liberación de mediadores inflamatorios como citocinas, quimiocinas, proteasas y radicales libres24. Las cito cinas secretadas por los macrófagos estimulan la expresión de MA en células endoteliales, lo que promueve el reclutamiento de más monocitos, creando un círculo vicioso que amplifica y perpetúa la reacción inflamatoria27 (fig. 2). El estudio detallado del papel de las citocinas en el desarrollo, progresión y complicaciones de la aterosclerosis se halla fuera del ámbito de este artículo, pero el lector interesado puede consultar una completa revisión sobre este tema recientemente publicada51. Entre las citocinas secretadas por los macrófagos destacan especialmente la interleucina (IL) 1, la IL-6 y el factor de necrosis tumoral a (TNF-a), ya que no sólo median respuestas inflamatorias locales, sino también efectos distantes, como la activación de genes hepáticos que codifican para proteínas de fase aguda como el fibrinógeno, el amiloide A sérico y la proteína C reactiva (PCR)52.
Aparte de su papel en la respuesta inflamatoria, estas citocinas actúan regulando la captación de lipoproteínas modificadas a través de los SR del macrófago y la salida o exportación de colesterol desde estas células. Así pues, la reducción de la expresión de citocinas en macrófagos puede ser una estrategia de importante valor terapéutico para reducir o ralentizar la progresión de la aterosclerosis. Sin embargo, el uso de antagonistas del TNF-a o de la IL-1 en clínica es muy limitado y se asocia con la aparición de importantes efectos adversos53. Una aproximación alternativa se basa en el hecho de que los PPAR y los receptores hepáticos X (LXR, liver X receptors) actúan como reguladores negativos de la respuesta inflamatoria en el macrófago; por tanto, PPAR y LXR constituyen dianas terapéuticas más adecuadas para modular la producción de citocinas en el macrófago54 (fig. 2).
En este sentido, se ha demostrado que los ligandos de PPARg inhiben la expresión de diversos genes proinflamatorios en macrófagos, entre ellos TNF-a e IL-1b55. El mecanismo molecular por el cual se producen estos efectos no está del todo claro; así, el agonista PPARg 15-deoxi prostaglandi na J2 inhibe la transcripción de citocinas mediada por NFkB por un mecanismo independiente de PPARg56. Además, la deleción de PPARg en macrófagos derivados de células madre no altera la secreción de citocinas57,58. Se ha propuesto que los efectos antiinflamatorios de agonistas PPARg como la rosiglitazona son independientes de PPARg sólo cuando el fármaco se usa a concentraciones mayores que su EC50; a estas concentraciones, el efecto antiinflamatorio podría atribuirse a la activación de PPARd59. De hecho, los agonistas PPARd suprimen la expresión de IL-1b en macrófagos, lo que indica que la activación de esta isoforma de PPAR es también antiinflamatoria60.
Se ha observado que los agonistas LXR reprimen una serie de genes proinflamatorios como la ciclooxigenasa 2, la IL-6 y la IL1b en macrófagos61. Dado que no se han identificado elementos de respuesta a LXR en las regiones promotoras de estos genes, se ha propuesto la transrepresión de NFkB por LXR como posible mecanismo de acción61. Aunque estos efectos parecen sugerir acciones beneficiosas de los agonistas LXR en la aterosclerosis, la realidad es que la utilización terapéutica de estos agentes plantea importantes problemas, co-mo se discutirá más adelante.
En cuanto a las estatinas, poseen propiedades antiinflamatorias que en parte son consecuencia de su capacidad de inhibir la adhesión de monocitos a la pared vascular y, por tanto, de reducir el número de células inflamatorias presentes en la lesión, como ya se ha comentado anteriormente. Pero además, las estatinas inhiben la producción de citocinas proinflamatorias en monocitos y macrófagos, lo que también contribuye a estos efectos. Así, estudios realizados en pacientes hipercolesterolémicos muestran que el tratamiento con simvastatina reduce la expresión de diversas citocinas (TNF, IL-1b62,63, IL-6 e IL-832). In vitro, la simvastatina, la atorvastatina y la cerivastatina inhiben la transcripción de citocinas inducida por el tratamiento con TNF-a en monocitos humanos32. El mecanismo está relacionado con la reducción de isoprenoides derivados de la vía del mevalonato, inhibición de la señalización por Rho y activación PPARg. La reducción en la expresión de citocinas inflamatorias por las estatinas no sólo se produce en macrófagos, sino también en otras células de la pared vascular (células musculares lisas, células endoteliales), lo que conduce a una menor concentración plasmática de estas citocinas y a una reducción de los valores de PCR, que se observa en diversos estudios clínicos realizados con este grupo de fármacos64-66.
Receptores scavenger y acumulación de colesterol en los macrófagos
Aparte de la inducción de TLR, la diferenciación de monocito a macrófago se caracteriza por el incremento en la expresión de receptores scavenger (SR). El término scavenger fue acuñado por Goldstein y Brown para distinguir estos receptores de los receptores de LDL nativas, cuya expresión está regulada por los valores intracelulares de esteroles67. Los SR, en cambio, no están sometidos a esta regulación, y debido aiacute;nas modificadas, contribuyen de forma importante a la acumulación de colesterol en los macrófagos67.
Los macrófagos expresan diversas clases de SR68,69, entre ellos el receptor scavenger de tipo A (SR-A), CD36, CD68, lectin-like oxidized receptor-1 (LOX-1) y el receptor scavenger que se une a fosfatidilserina y lipoproteínas oxidadas (SR-PSOX). La generación de ratones deficientes en SR-A y CD36 mostró que estos 2 receptores son responsables del 90% de la degradación de LDL acetiladas y oxidadas (ac-LDL y ox-LDL)70. Por ello, aunque otros receptores podrían contribuir a la acumulación de colesterol en el macrófago, por ejemplo CD6871, en la presente revisión nos referiremos únicamente a SR-A y CD36.
El gen correspondiente a SR-A, clonado en 1990 e inicialmente denominado receptor scavenger del macrófago (MSR)72, da lugar a 3 formas distintas del receptor por splicing alternativo del ARNm, denominados SR-AI, SR-AII y SR-AIII. Esta última forma contiene una alteración en la secuencia C-terminal y actúa como un mutante negativo de SR-A73. SR-AI y II se expresan en la superficie celular de macrófagos y células espumosas, y su presencia se ha detectado en placas ateroscleróticas74. Ambos receptores se unen a un amplio rango de moléculas polianiónicas, como las ac-LDL y las ox-LDL, aunque presentan una mayor afinidad por las prime-ras75. La función de SR-A en la captación de dichas lipoproteínas y la formación de células espumosas a partir de macrófagos sugiere, en principio, un papel proaterogénico para estos receptores. Para confirmar la implicación de SR-A en este proceso se generaron animales deficientes en estos receptores, y se cruzaron con modelos murinos susceptibles a la aterosclerosis. En los estudios iniciales realizados en ratones apo E/ 76 o LDLR/77, la eliminación de SR-A produjo una reducción en el tamaño de la lesión. Sin embargo, en un tercer estudio realizado en ratones hiperlipémicos apo E3 Leiden, la deleción de SR-A dio lugar a un incremento en el área de la lesión78. Estos resultados contradictorios se han atribuido a diferencias en la susceptibilidad a la aterosclerosis de las distintas cepas de ratones utilizadas. Sin embargo, estudios posteriores realizados en animales con una base genética más definida arrojaron también resultados confusos acerca del papel real de SR-A en la aterogénesis79,80. Además, la sobreexpresión de SR-A en los modelos murinos LDLR/81 o apo E/82 no dio lugar a diferencias significativas en el desarrollo de lesiones ateroscleróticas. Así pues, aunque resulta evidente que SR-A participa en la captación de lipoproteínas modificadas y en la formación de células espumosas, las discrepancias halladas en los diferentes modelos animales parecen indicar que este receptor presenta múltiples propiedades biológicas, algunas de las cuales son claramente proaterogénicas, mientras que otras podrían ser antiaterogénicas83. El predominio de uno u otro papel puede depender de factores locales o genéticos o estar relacionado con la fase de la enfermedad84. Así, el bloqueo de SR-A en las etapas iniciales del desarrollo de la aterosclerosis podría reducir la adherencia de macrófagos al endotelio y retrasar el desarrollo de lesiones85. Sin embargo, en fases más avanzadas la presencia de SR-A puede ser incluso beneficiosa, debido a su capacidad de inducir la apoptosis y la eliminación de células espumosas apoptóticas, reduciendo así la formación del núcleo necrótico de la lesión84.
En cuanto a CD36, se trata de un receptor scavenger de clase B implicado en múltiples procesos biológicos, incluyendo el metabolismo lipídico, la inflamación y la angiogénesis86. Su capacidad para reconocer e internalizar LDL, incluso mínimamente modificadas, parece implicarlo claramente en la formación de células espumosas. En este sentido, se ha observado que los macrófagos de pacientes que presentan un polimorfismo en el gen de CD3687, así como macrófagos procedentes de ratones deficientes en CD3688, muestran una menor capacidad de unir y captar ox-LDL. Además, los ratones doblemente deficientes en CD36 y apo E desarrollan menos lesiones ateroscleróticas que los correspondientes apo E/CD36+/+89. Resulta interesante que la exposición de macrófagos a ox-LDL da lugar a una marcada inducción de CD3690, mediada por la activación de PPARg por parte de los derivados oxidados de los ácidos grasos presentes en dichas lipoproteínas91,92. Esto implica que las ox-LDL inducen su propia captación por el receptor CD36 del macrófago, autoamplificando así el proceso de formación de células espumosas.
El papel de SR-A y CD36 como mediadores cruciales en este proceso ha sido de nuevo cuestionado en un trabajo recientemente publicado por Moore et al80, en el que se demuestra que la eliminación de uno u otro receptor no inhibe el desarrollo de aterosclerosis en ratones apo E/. Es posible que ello se deba a la expresión compensatoria de otros SR, o que vías alternativas de captación de lípidos independientes de la presencia de SR puedan suplir la carencia de SR-A y CD36. Por otra parte, la elimi-nación de estos genes puede afectar otras vías (eliminación de células apoptóticas, adhesión celular, defensa y respuesta inmune) que también condicionan el desarrollo de la aterosclerosis.
La diversidad de funciones biológicas atribuible a estos receptores dificulta el diseño de estrategias antiateroscleróticas basadas en modular terapéuticamente la actividad de SR-A y/o CD36. Sin embargo, diversos fármacos utilizados en clínica regulan la expresión de dichos receptores en macrófagos (fig. 3), efectos que pueden contribuir, en mayor o menor grado, a su eficacia terapéutica. Por ejemplo, se ha propuesto que las estatinas ejercen efectos directos sobre los macrófagos modulando la expresión de diversos SR por distintos mecanismos, no excluyentes entre sí: a) mediante la reducción de la oxidación de LDL, ya que las LDL modificadas inducen la expresión de los SR; b) mediante la inhibición de mediadores inflamatorios como NFkB, y c) como consecuencia de la inhibición de la isoprenilación de Rho y Ras. Se ha propuesto que las estatinas podrían actuar inhibiendo la expresión de SR-A93,94, aunque no todos los estudios realizados muestran dicha reducción95. De forma similar, los efectos de las estatinas sobre la expresión de CD36 en el macrófago tampoco están del todo claros, pues aunque en la mayoría de trabajos publicados se observa una reducción94-97, en ciertos estudios se evidencia un incremento98. Otro grupo de fármacos, las tiazolidindionas, produce una inducción de CD36 debido a la activación de PPARg, aunque este efecto es compensado por una reducción en la expresión de SR-A y por una activación de las vías de salida de colesterol del macrófago, por lo que no se traduce en un aumento de las concentraciones intracelulares de lípidos99.
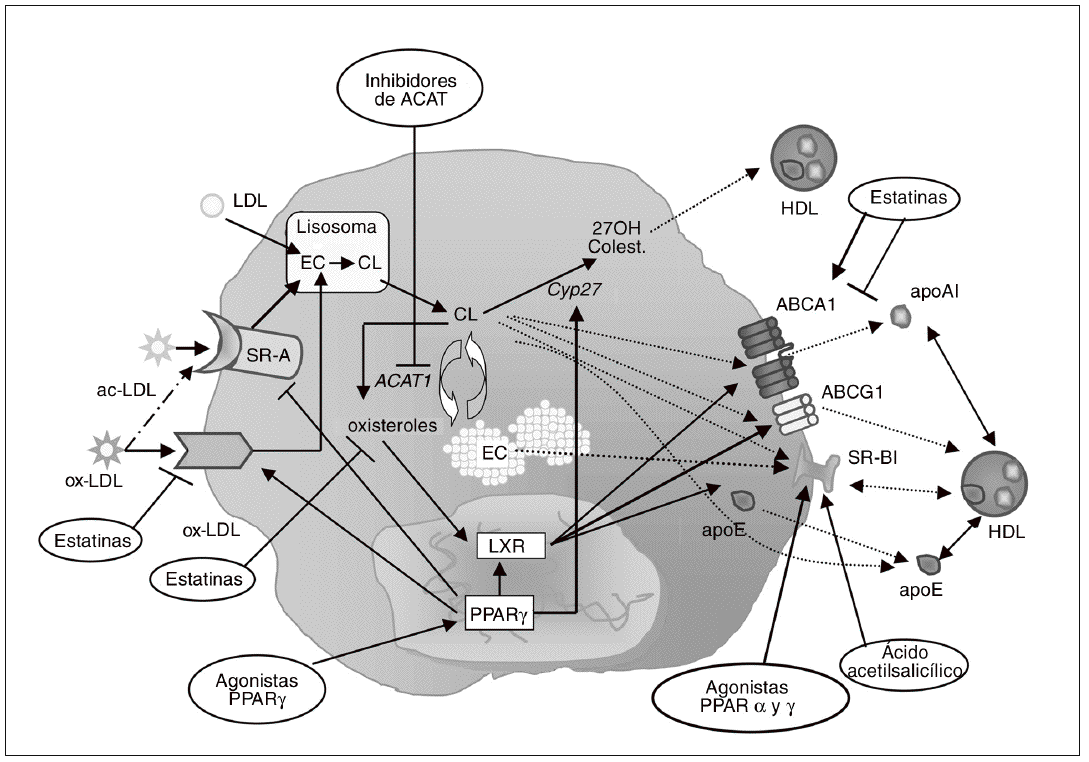
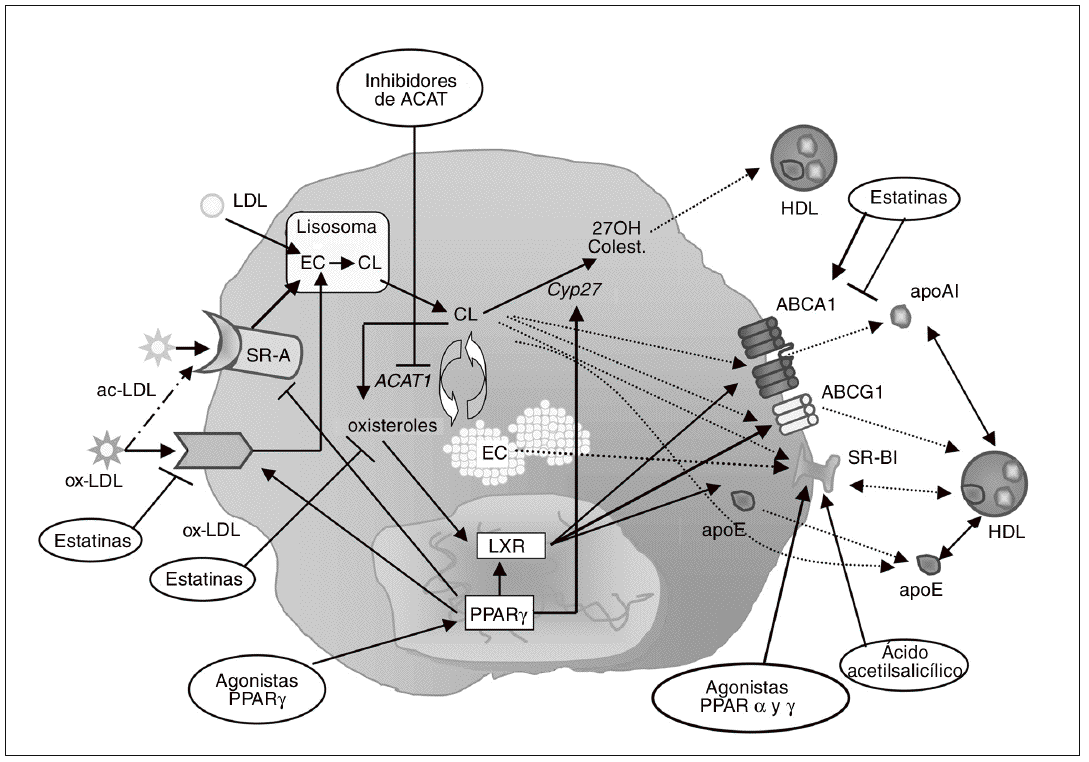
Figura 3. Inhibición de la formación de células espumosas como diana antiaterosclerótica. La diferenciación de monocito a macrófago se caracteriza por el incremento en la expresión de receptores scavenger, como el receptor scavenger de tipo A (SR-A) y CD36. Estos receptores contribuyen de forma muy importante a la acumulación de colesterol en los macrófagos, debido a su capacidad para unir e internalizar lipoproteínas modificadas como las LDL acetiladas (ac-LDL) u oxidadas (ox-LDL). Diversos agentes farmacológicos, como las estatinas y los agonistas PPARg, pueden regular la expresión de estos receptores en macrófagos. En el interior de estas células, los ésteres de colesterol (EC) captados con las lipoproteínas se hidrolizan formando colesterol libre (CL) y son reesterificados por acción de la enzima acil-CoA:colesterol aciltransferasa 1 (ACAT1). Los inhibidores de esta enzima, al bloquear la síntesis de EC para su almacenamiento en el macrófago, potencialmente reducen la formación de células espumosas. Por otra parte, el colesterol libre puede salir del macrófago/célula espumosa por distintos mecanismos: vehiculizado hacia las HDL maduras vía receptor scavenger BI (SR-BI) o transportador ABCG1, o hacia lipoproteínas poco lipidadas como la apo E o la apo A-I vía ABCA1. Otra posible vía de eflujo de colesterol implica su hidroxilación por la esterol 27-hidroxilasa (Cyp27) a metabolitos más polares que pueden ser vehiculizados hacia las HDL. Los LXR son los principales reguladores de muchos de los genes relacionados con la salida de colesterol del macrófago, como ABCA1, ABCG1 y apo E. Los agonistas PPARg inducen la expresión de ABCA1 y apoE en el macrófago a través de LXR. La estatinas pueden activar PPARg por su capacidad de reducir la geranilgeranilación de RhoA, y de este modo activar LXR e inducir la expresión génica de ABCA1; paradójicamente, al reducir la síntesis de oxiste roles, que son ligandos de LXR, las estatinas podrían reducir la activación de ABCA1.
Metabolismo intracelular de colesterol: papel de la acil-CoA:colesterol aciltransferasa
En el interior del macrófago, los ésteres de colesterol (EC) aportados por las lipoproteínas son hidrolizados y dan lugar a colesterol libre. Los reservorios celulares de colesterol esterificado y libre se hallan en un equilibrio dinámico controlado por las enzimas colesteril ester hidrolasa y acil-CoA:colesterol aciltransferasa (ACAT)100. Se han descrito 2 isoformas de esta enzima: ACAT1, la isoforma expresada en el macrófago101, sintetiza EC para su almacenamiento en el citosol celular, por lo que desempeña un papel crucial en la formación de células espumosas. De hecho, la ACAT1 se ha detectado en lesiones ateroscleróticas humanas, principalmente en células espumosas derivadas de macrófagos102. En cuanto a la ACAT2, se expresa principalmente en hepatocitos, donde actúa sintetizando los EC que serán secretados con las lipoproteínas de muy baja densidad (VLDL), y en células intestinales, en las que su actividad es necesaria para una correcta absorción del colesterol de la dieta101.
Debido a su implicación en el metabolismo lipoproteico y en la formación de células espumosas, la ACAT constituye una atractiva diana terapéutica para conseguir retrasar el desarrollo de la lesión aterosclerótica. Desde un punto de vista teórico, los inhibidores de ambas isoformas actuarían como fármacos hipolipemiantes y antiateroscleróticos a 3 niveles103: a) bloqueando la absorción intestinal de colesterol; b) inhibiendo la síntesis de VLDL hepática e incrementando el colesterol libre que puede ser excretado por la bilis, y c) inhibiendo la acumulación de EC en los macrófagos de la pared arterial, de forma que se reduciría la formación de células espumosas. Este último es un mecanismo antiaterosclerótico directo que podría bloquear el progreso de la lesión independientemente de cualquier efecto sobre los valores de lípidos plasmáticos. En este sentido, algunos inhibido res sistémicos de la ACAT, como el avasimibe o el HL-004, son capaces de reducir el almacenamiento de CE en macrófagos humanos104,105 y de inhibir el desarrollo de lesiones ateroscleróticas, e incluso inducir su regresión, en modelos animales106.
En las 2 últimas décadas se han desarrollado numerosos compuestos como inhibidores de la ACAT. Sin embargo, en las fases de investigación clínica dichos fármacos nunca han demostrado ser eficaces. Así, en el estudio A-PLUS107, en el que se administró avasimibe a más de 600 pacientes con enfermedad coronaria durante 2 años, se evidenció que el fármaco no reduce el volumen de la placa aterosclerótica, determinado por ultrasonografía intravascular; además se observó una tendencia hacia la elevación de los valores de colesterol LDL. Más recientemente, los efectos de otro inhibidor no selectivo de la ACAT, el pactimibe, se determinaron en 408 pacientes con aterosclerosis coronaria108. Los resultados también fueron negativos, ya que el volumen total del ateroma, así como el cambio de volumen en el segmento con la mayor gravedad de lesión, fueron significativamente peores en el grupo tratado con pactimibe que en el grupo placebo. A la luz de estos resultados, se deduce que la inhibición no selectiva de la ACAT es inefectiva como terapia antiaterosclerótica, e incluso puede resultar perjudicial. Los motivos de este aparente fracaso podrían deberse a que la inhibición de la actividad ACAT elimina un sistema de defensa del organismo contra la acumulación intracelular de colesterol libre. El exceso de colesterol libre que se produce al inhibir la ACAT no puede salir fácilmente de la célula y se acumula en su interior, causando toxicidad. Aunque es cierto que el colesterol sólo puede salir del macrófago cuando está en forma libre, también es cierto que para que se produzca una exportación efectiva de colesterol deben existir aceptores adecuados en el medio extracelular; por tanto, la inhibición de la ACAT debería combinarse con terapias que lograran incrementar la cifra de partículas aceptoras en el entorno de la placa, permitiendo así la salida del colesterol libre generado y su vehiculización hacia el hígado. Por el momento, el desarrollo terapéutico de inhibidores de la ACAT deberá realizarse con extrema cautela, teniendo en consideración estos posibles efectos adversos.
Salida del colesterol del macrófago
Las células periféricas dependen de la existencia de mecanismos de exportación de colesterol para mantener la homeostasis lipídica ya que, a diferencia de los hepatocitos, no disponen de sistemas de catabolismo como la formación de ácidos biliares. Esto es particularmente importante en el caso de los macrófagos, ya que, como se ha comentado anteriormente, la acumulación de colesterol no está controlada por mecanismos de regulación de la captación de lipoproteínas. Por ello, las vías de salida o exportación de colesterol constituyen dianas terapéuticas importantes en la prevención de la formación de células espumosas y del desarrollo de aterosclerosis109. Existen distintas vías para la salida de colesterol hacia el medio extracelular (fig. 3): a) difusión pasiva; b) transferencia de colesterol hacia apolipoproteínas pobres en lípidos, como la apo A-I, por medio del transportador ATP binding cassette 1 (ABCA1), y c) transporte de colesterol hacia lipoproteínas de alta densidad (HDL) maduras a través del transportador ABCG1 o del receptor scavenger SR-BI.
SR-BI es un receptor multiligando que media el flujo bidireccional entre las HDL y las células. El colesterol que sale del macrófago a través de SR-BI es convertido en EC por la enzima lecitina:colesterol aciltransferasa, y posteriormente estos EC que transportan las HDL pueden ser captados de forma selectiva por los hepatocitos a través del mismo receptor SR-BI. La participación de este receptor tanto en las fases iniciales como en las fases finales del proceso de transporte reverso de colesterol sugiere un rol protector contra el desarrollo de aterosclerosis110, lo que se ha confirmado en los estudios realizados en animales transgénicos. Así, la sobreexpresión hepática de SR-BI resulta en reducción de la aterosclerosis111-113, mientras que su eliminación en ratones apo E/ o LDLR/ induce el desarrollo de lesiones114,115. Incluso la eliminación selectiva de SR-BI en macrófagos causa un incremento en las lesiones115,116, lo que indica que el receptor SR-BI del macrófago es una diana potencial de terapia antiaterosclerótica (fig. 3). En este sentido, se ha demostrado que la activación de PPARa117 y g95,117,118 por agonistas específicos resulta en la inducción de CLA-1 (el homólogo humano de SR-BI) en monocitos humanos y macrófagos. Las estatinas son capaces también de inducir la expresión de SR-BI en macrófagos murinos119 y humanos118. Este efecto puede deberse a la inhibición de la respuesta inflamatoria, ya que NFkB modula la expresión de SR-BI119. El ácido acetilsalicílico es otro fármaco capaz de inducir la expresión de SR-BI en macrófagos humanos, probablemente por un mecanismo postranscripcional independiente de la inhibición de ciclooxigenasas120,121. Estos efectos sobre SR-BI podrán contribuir a las acciones ateroprotectoras que poseen los agonistas PPAR, las estatinas o el ácido acetilsalicílico.
Una de las rutas más importantes de exportación de colesterol del macrófago es la mediada por los transportadores ABCA1 y ABCG1 (fig. 3). El papel de ABCA1 en este proceso se descubrió al identificarlo como el gen defectuoso en la enfermedad de Tangier, caracterizada por una práctica ausencia de HDL y apo A-I en plasma y por la deposición de colesterol en los macrófagos tisulares122. A pesar de que no se cuestiona el papel fundamental de ABCA1 en el eflujo de colesterol, los experimentos realizados en ratones que sobreexpresan este transportador han dado lugar a resultados contradictorios en relación con el desarrollo de la aterosclerosis123,124; además, el estudio de ratones ABCA1/ no ha ayudado a resolver esta cuestión125. Sin embargo, en estudios de trasplante de médula ósea en los que el gen ABCA1 ha sido eliminado de forma selectiva de los macrófagos, se ha observado un incremento de lesiones ateroscleróticas126,127, y al contrario, el trasplante de macrófagos que sobreexpresan ABCA1 en ratones LDLR/ protege frente al desarrollo de aterosclerosis128. En cuanto a ABCG1, vehiculiza colesterol y fosfolípidos hacia las HDL129, un aceptor que se halla en mayor proporción que la apo A-I en plasma, por lo que su papel en el eflujo de colesterol podría ser incluso más relevante que el de ABCA1. Resulta interesante que la lipidación de la apo A-I mediada por ABCA1 da lugar a un aceptor eficiente del colesterol que es eliminado vía ABCG1, lo que sugiere que ambos transportadores actúan de forma sinérgica para facilitar el eflujo de colesterol desde el macrófago130. La expresión de ABCA1 y ABCG1 en macrófagos es controlada por los receptores nucleares LXRa y b131,132; se trata de un mecanismo homeostático diseñado para reducir la carga de colesterol libre intracelular, ya que se activa por los propios oxisteroles aportados por las lipoproteínas modificadas que el macrófago capta.
Aunque la importancia relativa y la relevancia fisiológica de ABCA1/G1 en el transporte reverso de colesterol desde el macrófago no están claramente establecidas, la modulación farmacológica de estos transportadores se considera una aproximación potencialmente beneficiosa para el tratamiento de las enfermedades cardiovasculares. En este aspecto, los agonistas PPARa y PPARg son capaces de inducir ABCA1 en macrófagos humanos, promoviendo así el eflujo de colesterol133. Paradójicamente, las estatinas parecen reducir la expresión de ABCA1 en diversos estudios in vitro118,134-137, lo que podría considerarse un efecto pleiotrópico negativo. Por el contrario, Argmann et al138 observaron que la atorvastatina aumenta los valores de ARNm de ABCA1 y ABCG1 y el eflujo de colesterol en macrófagos THP-1. Estos resultados divergentes se explicarían por un efecto dual de los intermediarios de la vía del mevalonato sobre ABCA1: por una parte, la depleción de isoprenoides producida por la estatina conduce a una menor geranilgeranilación de RhoA, con lo que se activaría PPARg y por consiguiente LXR, resultando en una inducción de ABCA1138; por otra parte, las estatinas reducen la síntesis de oxisteroles, que son ligandos de LXR, lo que conduciría a la menor activación de ABCA1136. De acuerdo con esta hipótesis, las estatinas no reducen la expresión de ABCA1 en presencia de un agonista LXR135, ni en macrófagos cargados de colesterol95,137. El hecho de que las estatinas no inhiban ABCA1 en macrófagos enriquecidos en colesterol sugiere que estos fármacos no afectarían negativamente el eflujo de colesterol en situaciones de hipercolesterolemia, de forma que sus propiedades antiaterogénicas no resultarían reducidas137.
La salida de colesterol hacia el medio extracelular es facilitada por la capacidad del propio macrófago de sintetizar y secretar apo E. Parece ser que la apo E secretada por los macrófagos puede actuar como aceptora de colesterol libre que sale de la célula en ausencia de otros aceptores en el medio139, de forma que facilita el transporte reverso de colesterol y contribuye a la reducción de la formación de células espumosas. El papel ateroprotector de la apo E parece confirmarse por experimentos en los que se demuestra que los macrófagos que no expresan apo E tienen menor capacidad de eflujo de colesterol140. Además, el trasplante de médula ósea de ratones deficientes en apo E a ratones LDLR/ aumenta el tamaño de las lesiones ateroscleróticas141. A la inversa, la expresión de la apo E en ratones apo E/ humana de forma selectiva en macrófagos es suficiente para reducir la aterosclerosis142. El mecanismo por el cual la secreción endógena de apo E estimula la salida de colesterol podría implicar, al menos en parte, el transportador ABCA1143. Así, se ha propuesto que la apo E secretada por el macrófago llega a la superficie celular en forma poco lipidada, donde es retenida por unión a proteoglucanos, facilitando la salida de lípidos a través de ABCA1144. Además, parece ser que ABCA1 modula la secreción de apo E por macrófagos humanos145. Sin embargo, el mecanismo no está del todo claro, ya que también existen evidencias que indican que la producción de apo E estimula el eflujo de colesterol independientemente de ABCA1143,146.
Gran parte de los genes que participan en el flujo de colesterol en los macrófagos están regulados por LXR, como es el caso de ABCA1, ABCG1131,132 y apo E147. Así, las tiazolidindionas incrementan la expresión de ABCA1 por un mecanismo que implica la activación de PPARg y la inducción de LXRa148. Los agonistas PPARg pueden actuar sobre otras vías de salida de colesterol de forma independiente de LXR, por ejemplo estimulando el eflujo hacia las HDL por inducción de ABCG1149. En cambio, la inhibición de la formación de células espumosas por parte de los agonistas PPARa parece requerir LXR, y posiblemente uno de los genes diana de LXR implicados es la apo E149. La regulación de la apo E es específica de tejido, ya que existe un elemento de respuesta a LXR en el promotor de la apo E de macrófagos y adipocitos pero no en células hepáticas147. De este modo, la activación LXR induce la salida de colesterol del macrófago no sólo por incremento de la expresión de ABCA1 sino también por el aumento de disponibilidad de aceptores del colesterol eliminado por esta vía, como la apo E. Además, la activación de LXR en macrófagos conduce a la inducción coordinada de otras apolipoproteínas, como la apo C-I, la apo C-IV y la apo C-II150.
Por todo lo que se ha expuesto, los LXR constituyen dianas muy atractivas para el desarrollo de fármacos antiateroscleróticos, ya que su activación tiene efectos antiinflamatorios, estimula el proceso de salida de colesterol del macrófago y el transporte reverso de colesterol in vivo151. En concordancia, los agonistas LXR reducen la formación de lesiones en modelos animales152. Sin embargo, la activación de LXR induce también la expresión la proteína de unión a elementos de respuesta a esteroles 1c (SREBP-1c)153, que controla la expresión de enzimas lipogénicas como la ácido graso sintasa (FAS). De forma similar a la inducción de transportadores ABC, el incremento en la expresión de FAS representa un mecanismo homeostático para reducir el potencial citotóxico del colesterol libre, en este caso promoviendo la síntesis de ácidos grasos para la formación de ésteres de colesterol. Sin embargo, este mecanismo adaptativo se convierte en un inconveniente cuando se trata de diseñar fármacos clínicamente útiles, ya que la activación LXR comportará esteatosis hepática e hiperlipemia. Se han propuesto diversas alternativas para evitar la aparición de estos efectos adversos: una de ellas es el diseño de agonistas selectivos de LXRb, ya que parece ser que el subtipo LXRa controla la expresi&oas que la isoforma b regula la expresión basal de ABCA1 en macrófagos154. De esta forma, la activación selectiva de LXRb podría estimular la salida de colesterol del macrófago sin inducir lipogénesis hepática. El principal problema de esta estrategia radica en la similitud entre los dominios de unión al ligando de una y otra isoforma, lo que dificulta encontrar agonistas específicos155. La segunda aproximación se basa en el hecho de que los genes diana de LXR difieren en su capacidad de reclutar coactivadores y correpresores. Por ejemplo, los heterodímeros LXR/RXR en ausencia de ligando reprimen la expresión de genes diana por unión a correpresores como NCoR, y la activación por ligando conduce a la disociación de NCoR y el reclutamiento de coactivadores. Sin embargo, en macrófagos LXR/ el correpresor NCoR no es reclutado a sus genes diana, lo cual es suficiente para permitir la inducción de ABCA1 sin que se desreprima SREBP1c ni se produzca un incremento en la síntesis de ácidos grasos54. Así pues, la síntesis de ligandos LXR que de forma selectiva impidieran la unión de correpresores sin inducir el reclutamiento de coactivadores resultaría en una activación LXR restringida a ciertos genes, evitándose el incremento en la lipogénesis.
El estudio de las vías metabólicas implicadas en la salida de colesterol de las células continúa desvelando nuevos mecanismos de regulación de este proceso. Así, la hidroxilación de colesterol por la enzima esterol 27-hidroxilasa (Cyp27) facilita su salida del macrófago al convertirlo en metabolitos más polares, como el 27-hidroxicolesterol156. Los macrófagos humanos expresan Cyp27 de forma particularmente abundante, probablemente como un mecanismo de defensa ante la acumulación de colesterol intracelular. De acuerdo con esta hipótesis, la expresión de Cyp27 está incrementada en tejidos ateroscleróticos, colocalizando con macrófagos en las lesiones más avanzadas157. Recientemente, Quinn et al158 han demostrado que los ligandos RXR y PPARg inducen la expresión de CYP27 en macrófagos, lo que podría constituir un nuevo mecanismo de protección contra el desarrollo de aterosclerosis mediado por estos agentes. Otro ejemplo lo constituye la proteína AEBP1 (adipocyte enhancer-binding protein 1), identificada como un nuevo regulador del eflujo de colesterol y de la respuesta inflamatoria en el macrófago159. Parece ser que AEBP1 reprime transcripcionalmente PPARg, LXRa y sus genes diana, induce la expresión de mediadores proinflamatorios y reduce la salida del colesterol del macrófago, promoviendo la formación de células espumosas. Por tanto, AEBP1 se suma a la lista de dianas farmacológicas potencialmente interesantes para el desarrollo de terapias antiaterogénicas.
Conclusión
En la actualidad la aterosclerosis ya no se contempla únicamente como un trastorno del metabolismo lipídico, sino como una enfermedad crónica de carácter inflamatorio en la que los monocitos y macrófagos desempeñan un papel muy destacado. En consecuencia, las aproximaciones farmacológicas destinadas a inhibir o retrasar el desarrollo de la aterosclerosis han pasado de la simple reducción de las concentraciones plasmáticas de lípidos a la intervención directa en las células de la pared arterial. Desde este punto de vista, la identificación de dianas terapéuticas en monocitos y macrófagos destinadas a reducir la adhesión y el reclutamiento de estas células, o a inhibir la inflamación y la formación de células espumosas, adquiere una especial relevancia. El desarrollo de nuevos fármacos que actúen modulando estas dianas puede llevar, en un futuro que no debería estar muy lejano, a una mejora sustancial en el tratamiento y la prevención de la enfermedad aterosclerótica.