







La hipercolesterolemia familiar homocigota (HFHo) es una enfermedad rara y potencialmente mortal que se caracteriza por la presencia en sangre de niveles muy elevados de colesterol en las lipoproteínas de baja densidad (cLDL) y por la aparición prematura y acelerada de enfermedad cardiovascular arteriosclerótica (ECVA). El Grupo de Consenso sobre hipercolesterolemia familiar de la Sociedad Europea de Arteriosclerosis (EAS) ha publicado recientemente una guía clínica para el diagnóstico y manejo clínico de la HFHo (Eur Heart J. 2014;35:2146-57).
Tanto la Sociedad Española de Arteriosclerosis (SEA) como la Fundación Hipercolesterolemia Familiar (FHF) consideran este documento de consenso europeo de gran valor y utilidad. Sin embargo, existen particularidades propias de nuestro país que aconsejan realizar una adaptación española del documento europeo de HFHo, con el objeto de aproximar esta útil guía de práctica clínica a nuestro entorno más próximo.
En España, el tratamiento crónico con estatinas, ezetimiba y resinas (colesevelam) tiene aportación reducida en el Sistema Nacional de Salud y es uno de los pocos países europeos donde la aféresis de LDL está incluida en la Cartera Básica de Servicios. El documento incluye, además, experiencias clínicas en el manejo de estos pacientes en nuestro país.
Este comité de redacción enfatiza la necesidad de un diagnóstico precoz de los pacientes con HFHo, su remisión inmediata a una unidad especializada y el inicio temprano del tratamiento apropiado. Se espera que estas recomendaciones constituyan una guía para el abordaje del paciente con HFHo en España.
Homozygous familial hypercholesterolaemia (HoFH) is a rare life-threatening disease characterized by markedly elevated circulating levels of low-density lipoprotein cholesterol (LDL-C) and accelerated, premature atherosclerotic cardiovascular disease (ACVD).
The Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society (EAS) has recently published a clinical guide to diagnose and manage HoFH (Eur Heart J. 2014;35:2146-57).
Both the Spanish Society of Atherosclerosis (SEA) and Familial Hypercholesterolaemia Foundation (FHF) consider this European Consensus document of great value and utility. However, there are particularities in our country which advise to have a Spanish adaptation of the European HoFH document in order to approximate this clinical guide to our environment.
In Spain, chronic treatment with statins, ezetimibe and resins (colesevelam) has a reduced contribution in the National Health System (NHS) and is one of the few European countries where LDL apheresis is included in the Basic Service Portfolio coverage. This Spanish document also includes clinical experience in the management of these patients in our country.
The Drafting Committee emphasizes the need for early identification of HoFH patients, prompt referral to specialized units, and an early and appropriate treatment. These recommendations will provide a guidance for HoFH patient management in Spain.
La hipercolesterolemia familiar homocigota (HFHo) es una enfermedad rara y potencialmente mortal que se caracteriza clínicamente, por la presencia de niveles plasmáticos de colesterol total >500mg/dl (>13mmol/l), xantomas extensos y enfermedad cardiovascular arteriosclerótica (ECVA) prematura y progresiva. Los estudios experimentales han demostrado un grave defecto de la capacidad de unión e internalización de las partículas de LDL, generalmente causado por mutaciones en los 2 alelos del gen que codifica el receptor de LDL (LDLR)1. Además, algunos pacientes con HFHo pueden presentar mutaciones en otros genes, como APOB, PCSK9 y LDLRAP1, también implicados en la captación de LDL.
Si no reciben tratamiento, la mayoría de estos pacientes desarrollan arteriosclerosis sintomática antes de los 20años de edad y generalmente fallecen antes de los 30años1. Por esta razón, los objetivos principales del tratamiento son la prevención de la ECVA mediante el control precoz y agresivo de la hipercolesterolemia y la detección temprana de las complicaciones vasculares, de las que cabe destacar la oclusión del ostium coronario, la estenosis aórtica y la alteración de las coronarias2. Desafortunadamente, la HFHo normalmente se diagnostica cuando ya se ha desarrollado una arteriosclerosis coronaria importante, lo que pone de manifiesto la necesidad de optimizar el diagnóstico y tratamiento en niños.
El Grupo de Consenso sobre hipercolesterolemia familiar (HF) de la Sociedad Europea de Arteriosclerosis (EAS) ha generado una guía clínica para la detección y tratamiento de los pacientes con HFHo en Europa, siendo un documento de referencia a la hora de tomar decisiones sobre la atención médica que debe prestarse a estos pacientes (Cuchel et al.3).
Tanto la Sociedad Española de Arteriosclerosis (SEA) como la Fundación Hipercolesterolemia Familiar (FHF) consideran este documento de gran valor y utilidad en nuestro país. Anteriormente no existían guías de práctica clínica exclusivas de HFHo, estando dicha enfermedad incluida de manera somera dentro de guías de manejo más generales4,5.
Sin embargo, existen particularidades propias de nuestro país, como son los aspectos epidemiológicos y diagnósticos, la oferta de procedimientos terapéuticos, la sensibilización del sistema sanitario a las necesidades del tratamiento crónico de esta grave enfermedad genética (aportación reducida) y la experiencia clínica en el manejo de pacientes con HFHo. Esta es la razón por la cual este Comité considera conveniente realizar una adaptación española del documento europeo de HFHo, con el objeto de aproximar esta guía de práctica clínica a nuestro entorno más próximo.
Estas recomendaciones están dirigidas a un amplio espectro de médicos (cardiólogos y responsables de unidades de lípidos), pero de forma muy especial a aquellos de otras especialidades (atención primaria, pediatras y dermatólogos) susceptibles de sospechar y detectar por primera vez este tipo de pacientes, pudiendo remitirlos a las unidades especializadas de forma precoz.
La SEA y la FHF han encargado a este comité de redacción la adaptación de estas guías europeas a la realidad de nuestro país.
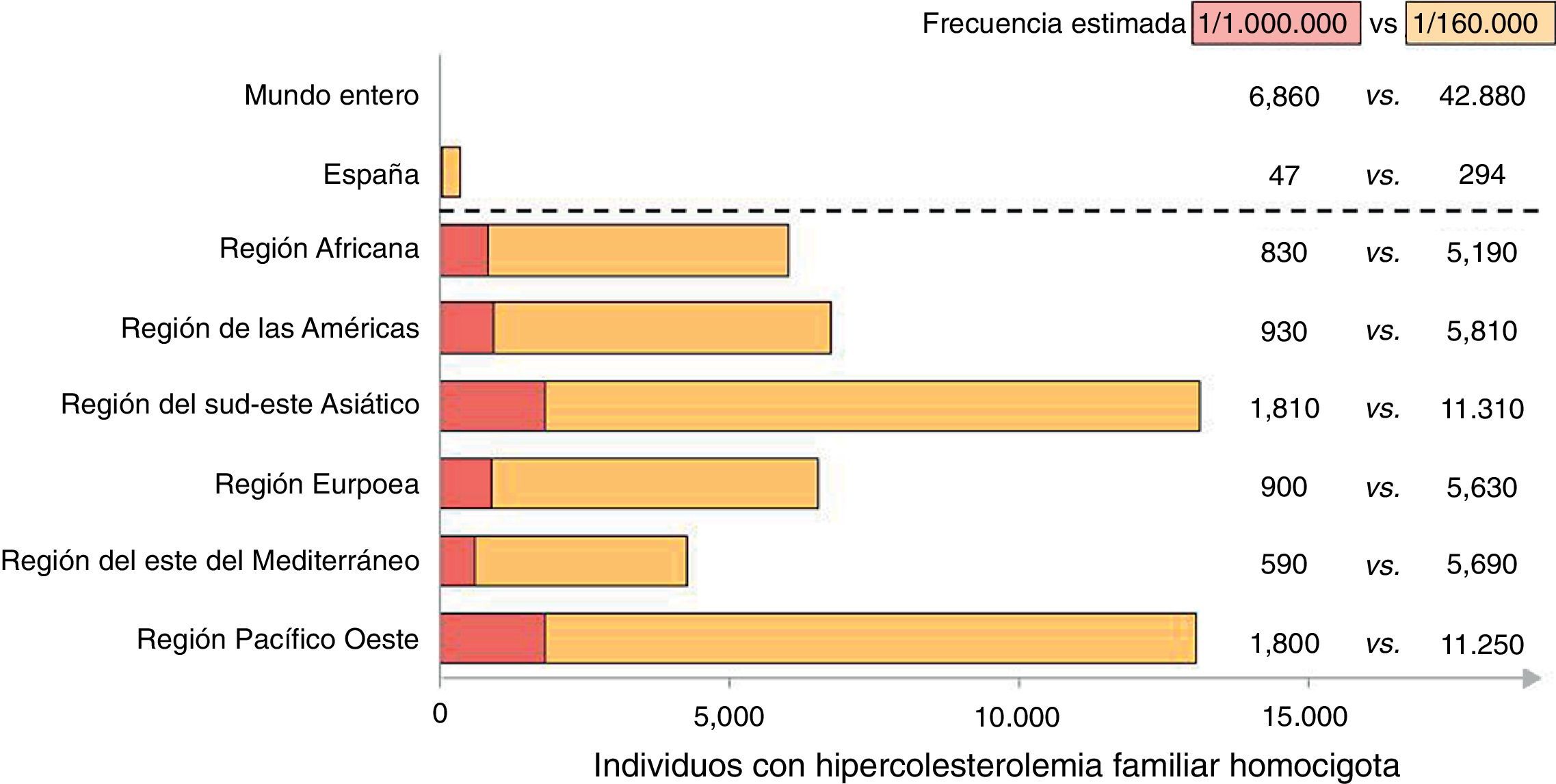
Prevalencia de la hipercolesterolemia familiar homocigota clínica y confirmada genéticamenteHistóricamente, la frecuencia de la HFHo clínica se ha venido estimando en una de cada 1.000.000 personas, y la frecuencia de la HF heterocigota (HFHe), en una de cada 500personas1, aunque se han descrito frecuencias más elevadas en otras poblaciones, como los francocanadienses, los de Sudáfrica o los libaneses cristianos, debido al efecto fundador por aislamiento genético6. Este valor histórico de prevalencia de HFHo podría estar infraestimado, puesto que únicamente se refiere a los pacientes con la misma mutación en el receptor LDL7. Estudios recientes realizados en población general sugieren que, según los criterios de la Red de Clínicas de Lípidos de Holanda, la prevalencia de la HFHe sería mayor, una de cada ∼200 personas8 o, en el caso de la HFHe definida molecularmente, una de cada 244personas9. En consecuencia, la HFHo podría afectar entre una de cada 160.000 a 300.000 personas (fig. 1).
, así como en las estimaciones realizadas directamente sobre la población general danesa (∼1/160.000).Datos de Nordestgaard et al.8.")
Número estimado de individuos en todo el mundo con HFHo clasificados según las regiones de la Organización Mundial de la Salud y de forma individual, España. Las estimaciones se basan en los datos históricos de prevalencia (uno entre un millón con HFHo), así como en las estimaciones realizadas directamente sobre la población general danesa (∼1/160.000).Datos de Nordestgaard et al.8.
En España no existen estudios publicados que arrojen datos sobre la prevalencia nacional de la HFHo. Sin embargo, un estudio multicéntrico llevado a cabo en 17 comunidades autónomas españolas para la determinación genética en cerca de 7.700 pacientes con HF (5.430 de los cuales eran casos índice) detectó 13 pacientes homocigotos verdaderos y 17 heterocigotos compuestos. Además, 2.223 familiares de casos positivos fueron también cribados, detectando 4 pacientes homocigotos más. En total, el estudio de Palacios et al.10 confirmó genéticamente 34 pacientes con HFHo en España hasta el año 2011. En los últimos 3años, se han identificado al menos 10 nuevos casos confirmados genéticamente (comunicación personal de miembros del Comité).
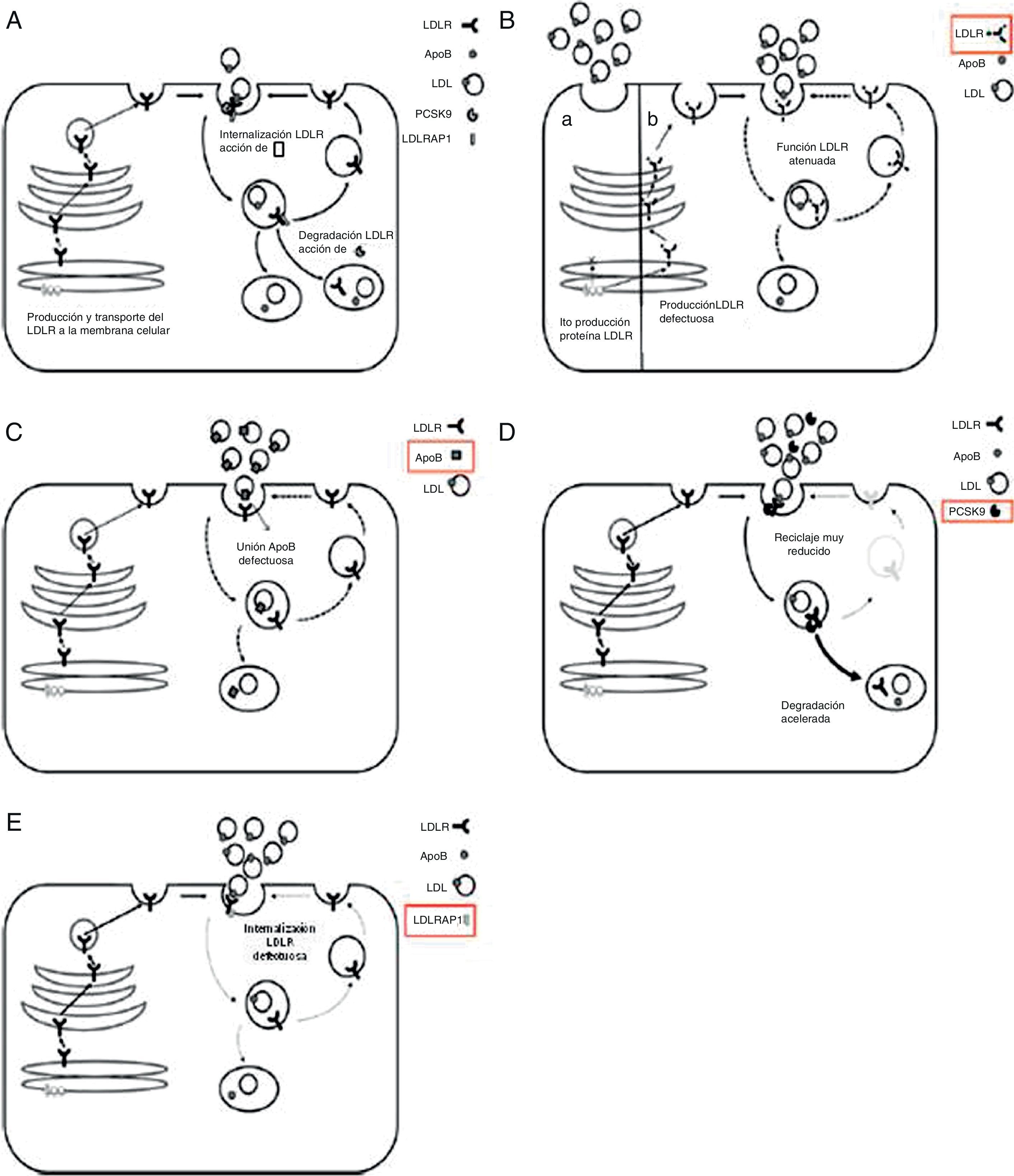
Genética de la hipercolesterolemia familiar homocigotaEn la figura 2 se resumen las proteínas conocidas que afectan a la función del receptor de LDL, así como el papel que desempeña cada una de ellas. La mayoría de los pacientes con HFHo confirmada genéticamente tienen 2 alelos mutados del gen LDLR (>95%; MIM606945) y cada uno de sus progenitores presenta HFHe. Recientemente se han identificado mutaciones en alelos de otros 3 genes como causantes de casos con un fenotipo grave de HFHo. Estos genes secundarios son APOB (2-5%; MIM107730), que codifica la apolipoproteína (apo) B; PCSK9 (<1%; MIM607786), que codifica la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9), y LDLRAP1 (<1%; MIM695747), que codifica la proteína adaptadora1 del receptor de LDL, que causa un fenotipo recesivo, ya que los progenitores portadores presentan perfiles lipídicos normales11. Los pacientes son homocigotos, con la misma mutación en ambos alelos del mismo gen o, más a menudo, heterocigotos compuestos con mutaciones diferentes en cada alelo del mismo gen, o heterocigotos dobles con mutaciones en 2 genes diferentes que afectan a la función del receptor de LDL (fig. 3).
 1. El receptor de lipoproteínas de baja densidad (LDLR) recién sintetizado es transportado a la membrana celular. Tras alcanzar la superficie celular, el receptor de lipoproteínas de baja densidad se une a la apolipoproteína B-100 (apoB-100), la proteína principal de las LDL, formando un complejo. 2. El complejo receptor de lipoproteínas de baja densidad-lipoproteína de baja densidad, localizado en una invaginación recubierta de clatrina, es endocitado mediante interacciones que incluyen a la proteína adaptadora 1 del receptor de lipoproteínas de baja densidad (LDLRAP1). 3. Dentro del endosoma, el complejo se disocia: apoB-100 y lípidos se dirigen a los lisosomas, donde se degradan; el receptor de lipoproteínas de baja densidad se recicla a la membrana celular. 4. La proproteína convertasa subtilisina/kexina de tipo 9 (PCSK9) actúa como inhibidor postranscripcional del receptor de lipoproteínas de baja densidad y, a través de una interacción con él, dirige al receptor de lipoproteínas de baja densidad hacia la degradación en lugar de hacia el reciclaje. B) En presencia de mutaciones de pérdida de función en el gen que codifica el receptor de lipoproteínas de baja densidad, este receptor no se sintetiza, no se transporta ni se presenta en la superficie, por lo que su función está alterada. C) En presencia de mutaciones en la región de unión de la apoB al receptor de lipoproteínas de baja densidad, disminuye su capacidad para unirse al receptor de lipoproteínas de baja densidad, con la consecuente reducción de la captación de partículas de lipoproteínas de baja densidad. D) En presencia de mutaciones de aumento de función en el gen que codifica la PCSK9, la mayoría de los receptores de lipoproteínas de baja densidad se dirige a la degradación, con la consiguiente reducción del número de receptores de lipoproteínas de baja densidad que se reciclan a la superficie celular. E) En presencia de mutaciones de pérdida de función en el gen que codifica la LDLRAP1, que facilita la interacción entre el receptor de lipoproteínas de baja densidad y la maquinaria celular que regula el proceso de endocitosis, se altera la internalización del complejo receptor de lipoproteínas de baja densidad-proteínas de baja densidad.")
Proteínas que afectan a la función del receptor de lipoproteínas de baja densidad. A) 1. El receptor de lipoproteínas de baja densidad (LDLR) recién sintetizado es transportado a la membrana celular. Tras alcanzar la superficie celular, el receptor de lipoproteínas de baja densidad se une a la apolipoproteína B-100 (apoB-100), la proteína principal de las LDL, formando un complejo. 2. El complejo receptor de lipoproteínas de baja densidad-lipoproteína de baja densidad, localizado en una invaginación recubierta de clatrina, es endocitado mediante interacciones que incluyen a la proteína adaptadora 1 del receptor de lipoproteínas de baja densidad (LDLRAP1). 3. Dentro del endosoma, el complejo se disocia: apoB-100 y lípidos se dirigen a los lisosomas, donde se degradan; el receptor de lipoproteínas de baja densidad se recicla a la membrana celular. 4. La proproteína convertasa subtilisina/kexina de tipo 9 (PCSK9) actúa como inhibidor postranscripcional del receptor de lipoproteínas de baja densidad y, a través de una interacción con él, dirige al receptor de lipoproteínas de baja densidad hacia la degradación en lugar de hacia el reciclaje. B) En presencia de mutaciones de pérdida de función en el gen que codifica el receptor de lipoproteínas de baja densidad, este receptor no se sintetiza, no se transporta ni se presenta en la superficie, por lo que su función está alterada. C) En presencia de mutaciones en la región de unión de la apoB al receptor de lipoproteínas de baja densidad, disminuye su capacidad para unirse al receptor de lipoproteínas de baja densidad, con la consecuente reducción de la captación de partículas de lipoproteínas de baja densidad. D) En presencia de mutaciones de aumento de función en el gen que codifica la PCSK9, la mayoría de los receptores de lipoproteínas de baja densidad se dirige a la degradación, con la consiguiente reducción del número de receptores de lipoproteínas de baja densidad que se reciclan a la superficie celular. E) En presencia de mutaciones de pérdida de función en el gen que codifica la LDLRAP1, que facilita la interacción entre el receptor de lipoproteínas de baja densidad y la maquinaria celular que regula el proceso de endocitosis, se altera la internalización del complejo receptor de lipoproteínas de baja densidad-proteínas de baja densidad.
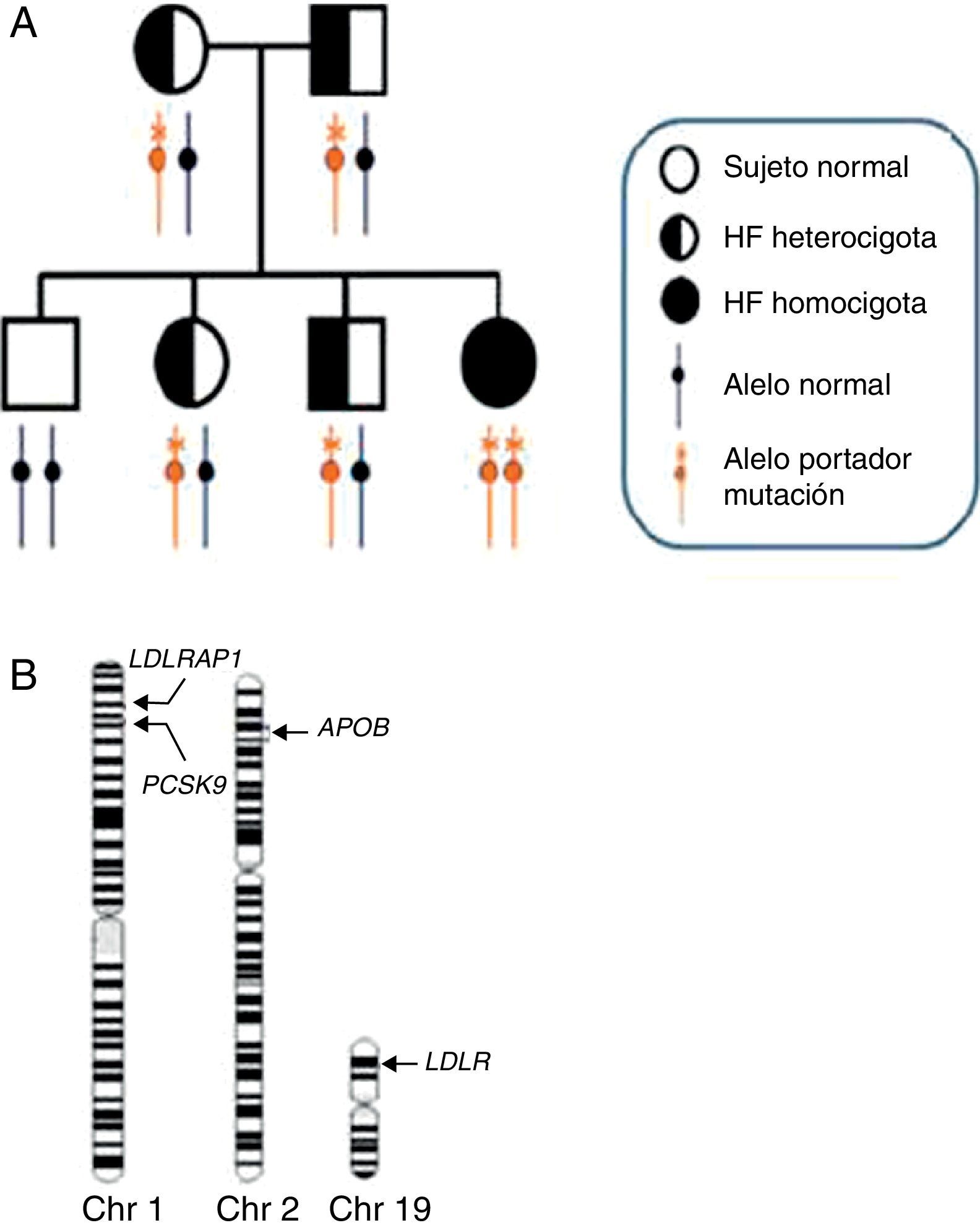
 Herencia de la hipercolesterolemia familiar homocigota en un árbol genealógico. En un emparejamiento entre padres heterocigotos, cada uno de ellos portador de una copia de un alelo con la mutación para la hipercolesterolemia familiar, el 25% de los hijos tendrán 2 copias de los alelos normales (homocigoto normal), el 50% serán heterocigotos y el 25% restante serán portadores de 2 copias de alelos con la mutación para la hipercolesterolemia familiar (HFHo). Los genes y tipos de mutación específicos que se heredan determinan si el individuo afectado es un homocigoto simple o un heterocigoto compuesto o doble. B) Heterogeneidad genética de la hipercolesterolemia familiar. En los ideogramas de los cromosomas 1, 2 y 19 se indican las posiciones de los 4 principales genes causantes de la hipercolesterolemia familiar, que por orden descendiente de frecuencia son LDLR (>95%), APOB (2-5%), PCSK9 (<1%) y LDLRAP1 (<1%). En la gran mayoría de los pacientes con HFHo representados en (A), los alelos causantes de la mutación están dentro del mismo gen (normalmente LDLR); estos pacientes se definen como «homocigotos puros». Los pacientes con HFHo portadores de la misma mutación en cada alelo se denominan «homocigotos simples», mientras que los que heredan diferentes mutaciones dentro del mismo gen se denominan «heterocigotos compuestos». Finalmente, existen casos muy raros de pacientes con hipercolesterolemia familiar que presentan alelos portadores de mutaciones para la hipercolesterolemia familiar de 2 loci de hipercolesterolemia familiar diferentes: la primera está prácticamente siempre en el gen LDLR, mientras que la otra está en uno de los otros 3 loci. Estos pacientes se denominan «heterocigotos dobles».")
Genética y heterogeneidad genética de la HFHo. A) Herencia de la hipercolesterolemia familiar homocigota en un árbol genealógico. En un emparejamiento entre padres heterocigotos, cada uno de ellos portador de una copia de un alelo con la mutación para la hipercolesterolemia familiar, el 25% de los hijos tendrán 2 copias de los alelos normales (homocigoto normal), el 50% serán heterocigotos y el 25% restante serán portadores de 2 copias de alelos con la mutación para la hipercolesterolemia familiar (HFHo). Los genes y tipos de mutación específicos que se heredan determinan si el individuo afectado es un homocigoto simple o un heterocigoto compuesto o doble. B) Heterogeneidad genética de la hipercolesterolemia familiar. En los ideogramas de los cromosomas 1, 2 y 19 se indican las posiciones de los 4 principales genes causantes de la hipercolesterolemia familiar, que por orden descendiente de frecuencia son LDLR (>95%), APOB (2-5%), PCSK9 (<1%) y LDLRAP1 (<1%). En la gran mayoría de los pacientes con HFHo representados en (A), los alelos causantes de la mutación están dentro del mismo gen (normalmente LDLR); estos pacientes se definen como «homocigotos puros». Los pacientes con HFHo portadores de la misma mutación en cada alelo se denominan «homocigotos simples», mientras que los que heredan diferentes mutaciones dentro del mismo gen se denominan «heterocigotos compuestos». Finalmente, existen casos muy raros de pacientes con hipercolesterolemia familiar que presentan alelos portadores de mutaciones para la hipercolesterolemia familiar de 2 loci de hipercolesterolemia familiar diferentes: la primera está prácticamente siempre en el gen LDLR, mientras que la otra está en uno de los otros 3 loci. Estos pacientes se denominan «heterocigotos dobles».
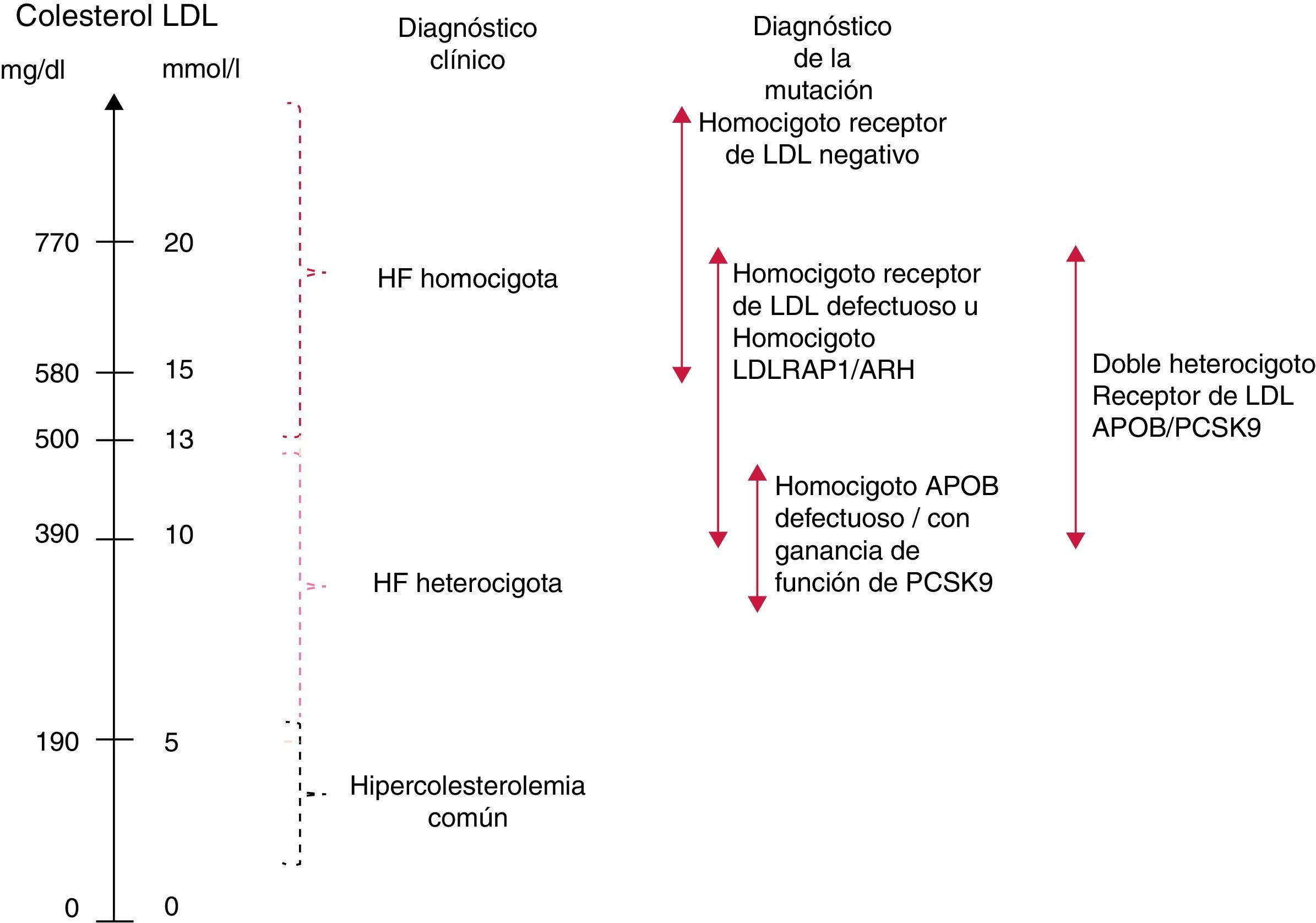
Independientemente del defecto genético subyacente, la gravedad del fenotipo HFHo depende de la actividad residual del receptor de LDL. Según los ensayos in vitro realizados con los fibroblastos cultivados, los pacientes con HFHo definida clínicamente se han clasificado convencionalmente como pacientes con receptor negativo (actividad residual <2%) o con receptor defectuoso (actividad residual 2-25%)1. Los pacientes con HFHo que son LDLR negativo tienen niveles más altos de colesterol ligado a las lipoproteínas de baja densidad (cLDL) y un pronóstico clínico más desfavorable que los pacientes con LDLR defectuoso2,12,13.
No se ha evaluado sistemáticamente la actividad residual del receptor de LDL en pacientes portadores de mutaciones en los genes APOB y PCSK9. En pacientes portadores de mutaciones en LDLRAP1, la actividad del receptor de LDL en el cultivo de fibroblastos es normal, por lo que la causa de la hipercolesterolemia sigue sin estar clara11,14. No obstante, los nuevos datos sugieren que los portadores de mutaciones en estos genes pueden presentar un fenotipo más leve que los sujetos negativos para el receptor11. En general, los niveles medios de cLDL por genotipo aumentan en el siguiente orden: HFHe <heterocigoto doble (p.ej., LDLR +mutación de ganancia de función en PCSK9 o mutación en APOB) <homocigoto para una mutación de ganancia de función en PCSK9 o APOB <homocigoto para mutaciones de LDLR defectuoso o LDLRAP1 <heterocigoto compuesto para mutación de LDLR defectuoso +mutación de LDLR negativo <homocigoto para mutaciones de LDLR negativo (véase la fig. 4 y el material complementario del documento de Consenso Europeo, disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4139706/bin/supp_35_32_2146__index.html).
; PCSK9: proproteína convertasa subtilisina/kexina tipo 9.")
Variabilidad fenotípica en la HFHo. En Internet se puede consultar el material complementario si se desea obtener la explicación detallada y la bibliografía relevante.APOB: apolipoproteína B; LDL: lipoproteína de baja densidad; LDLRAP1: proteína adaptadora 1 del receptor de LDL (es decir, ARH, hipercolesterolemia autosómica recesiva); PCSK9: proproteína convertasa subtilisina/kexina tipo 9.
Otras fuentes de variabilidad en el fenotipo de HFHo pueden surgir a partir de variantes genéticas comunes de pequeño efecto (polimorfismos comunes de un solo nucleótido), interacciones entre genes y entre genes-ambiente, así como influencias no mendelianas y epigenéticas11,15,16. Un mayor conocimiento del genoma y las nuevas técnicas de secuenciación son fundamentales para definir dicha variabilidad, así como los genes causantes o situaciones adicionales modificantes, que tienen o tendrán importantes implicaciones pronósticas y terapéuticas.
Características metabólicas de la hipercolesterolemia familiar homocigotaEn la HFHo, la causa subyacente de la grave hipercolesterolemia está en la alteración del funcionamiento del receptor de LDL. Aunque su principal y más directo efecto es la captación hepática defectuosa de LDL, hay otras alteraciones metabólicas que pueden contribuir a las características metabólicas y a la enfermedad arteriosclerótica acelerada asociada a la HFHo. El metabolismo de la apoB en la HFHo sigue sin ser completamente conocido, aunque los estudios in vitro e in vivo sugieren que las mutaciones de LDLR negativo también se asocian al aumento de la secreción hepática de apoB. Asimismo, aunque frecuentemente los niveles de triglicéridos están dentro del intervalo normal, se puede observar hipertrigliceridemia que suele ser secundaria a la obesidad, o al síndrome metabólico de alta prevalencia, aunque también pudiera tener un papel la reducción del catabolismo de lipoproteínas ricas en triglicéridos a consecuencia de la función deficiente del receptor de LDL. La HF también se asocia a un aumento de los niveles plasmáticos de lipoproteína(a) [Lp(a)] a través de un mecanismo no definido que puede no afectar directamente a la vía del receptor de LDL. La Lp(a) es un potente e independiente factor de riesgo para los episodios coronarios en la HFHe y se debe determinar su concentración plasmática. Si esta es >50mg/dl independientemente de las concentraciones de cLDL, el paciente es de riesgo muy alto y el tratamiento de los pacientes con HFHe debe ser más intenso17. Los niveles de Lp(a) tienden a ser más altos en la HFHo que en la HFHe y son independientes de la variación del gen APOA8. Por último, los pacientes con HFHo a menudo presentan niveles bajos de colesterol unido a lipoproteína de alta densidad (cHDL), probablemente debido al recambio acelerado de la apoA-1 de HDL y un flujo de salida defectuoso del colesterol unido a HDL. Estas alteraciones metabólicas han sido ampliamente estudiadas18.
Diagnóstico de la hipercolesterolemia familiar homocigotaEl diagnóstico de la HFHo se puede realizar en función de criterios genéticos y/o clínicos (tabla 1). Aunque el análisis genético puede proporcionar un diagnóstico definitivo de la HFHo, se reconoce que en algunos pacientes sigue siendo difícil lograr la confirmación genética a pesar de investigaciones exhaustivas; de hecho, no se puede excluir la existencia de genes de HF adicionales8. Históricamente la HFHo se ha diagnosticado teniendo en cuenta una concentración plasmática de cLDL sin tratamiento ≥500mg/dl (≥13mmol/l) o una concentración de cLDL con tratamiento ≥300mg/dl (≥8mmol/l) (estos niveles de cLDL son solo indicativos, y niveles más bajos, especialmente en niños o en pacientes tratados, no excluyen HFHo), y la presencia de xantomas cutáneos y/o tendinosos antes de los 10años de edad, o la presencia de niveles elevados de cLDL sin tratamiento en ambos progenitores, compatible con HFHe.
Criterios para el diagnóstico de la hipercolesterolemia familiar homocigota
| • Confirmación genética de 2 alelos portadores de mutaciones funcionales en el locus del gen LDLR, APOB, PCSK9 o LDLRAP1O |
| • cLDL sin tratamiento ≥500mg/dl (≥13mmol/l) o cLDL con tratamiento ≥300mg/dl (≥8mmol/l) (estos niveles de cLDL son solo indicativos, y niveles más bajos, especialmente en niños o en pacientes tratados, no excluyen HFHo), junto con: |
| – Xantomas cutáneos y/o tendinosos evidenciados antes de los 10años de edad, o |
| – Niveles elevados de cLDL sin tratamiento en ambos progenitores, compatible con HFHe |
Dentro de una misma familia, el nivel de cLDL en plasma es un discriminador fundamental, siendo aproximadamente 4 veces más alto en los miembros con HFHo y 2 veces más alto en los miembros con HFHe, en comparación con los no afectados11. A nivel poblacional, sin embargo, el intervalo de los niveles de cLDL puede solaparse significativamente entre la HFHe y la HFHo (fig. 4), y se pueden encontrar niveles de cLDL sin tratamiento <500mg/dl (<13mmol/l) en la HFHo genéticamente confirmada9,13. Esto es especialmente importante en el caso de los niños, que tienden a presentar niveles de cLDL inferiores a los de los adultos. Más del 50% de los niños con HFHo identificados en los Países Bajos tienen niveles de cLDL comprendidos entre 217mg/dl (5,6mmol/l) y 379mg/dl (9,8mmol/l) (comunicación personal de A. Wiegman). Esta heterogeneidad fenotípica se puede explicar al menos parcialmente por la heterogeneidad genotípica descrita previamente. Por ello, los límites de cLDL que se indican aquí deben tomarse exclusivamente como una guía para el diagnóstico. De hecho, el límite de cLDL con tratamiento de >300mg/dl (>8mmol/l) se considera hoy en día obsoleto, dada la gran variedad de tratamientos hipolipemiantes que reciben normalmente estos pacientes. Este punto se ilustra con claridad en un reciente ensayo clínico en el que pacientes con HFHo con un diagnóstico genético confirmado presentaban niveles basales de cLDL de solo ∼150mg/dl (3,9mmol/l) mientras estaban en tratamiento con varios agentes reductores de cLDL19; lo mismo puede verse en una publicación reciente9. Por todo lo anterior, en los pacientes con concentraciones de cLDL superiores a 200mg/dl después de tratamiento con estatinas potentes a dosis altas o tratamiento combinado se debe descartar genéticamente HFHo, sobre todo si existe hipercolesterolemia en ambos progenitores.
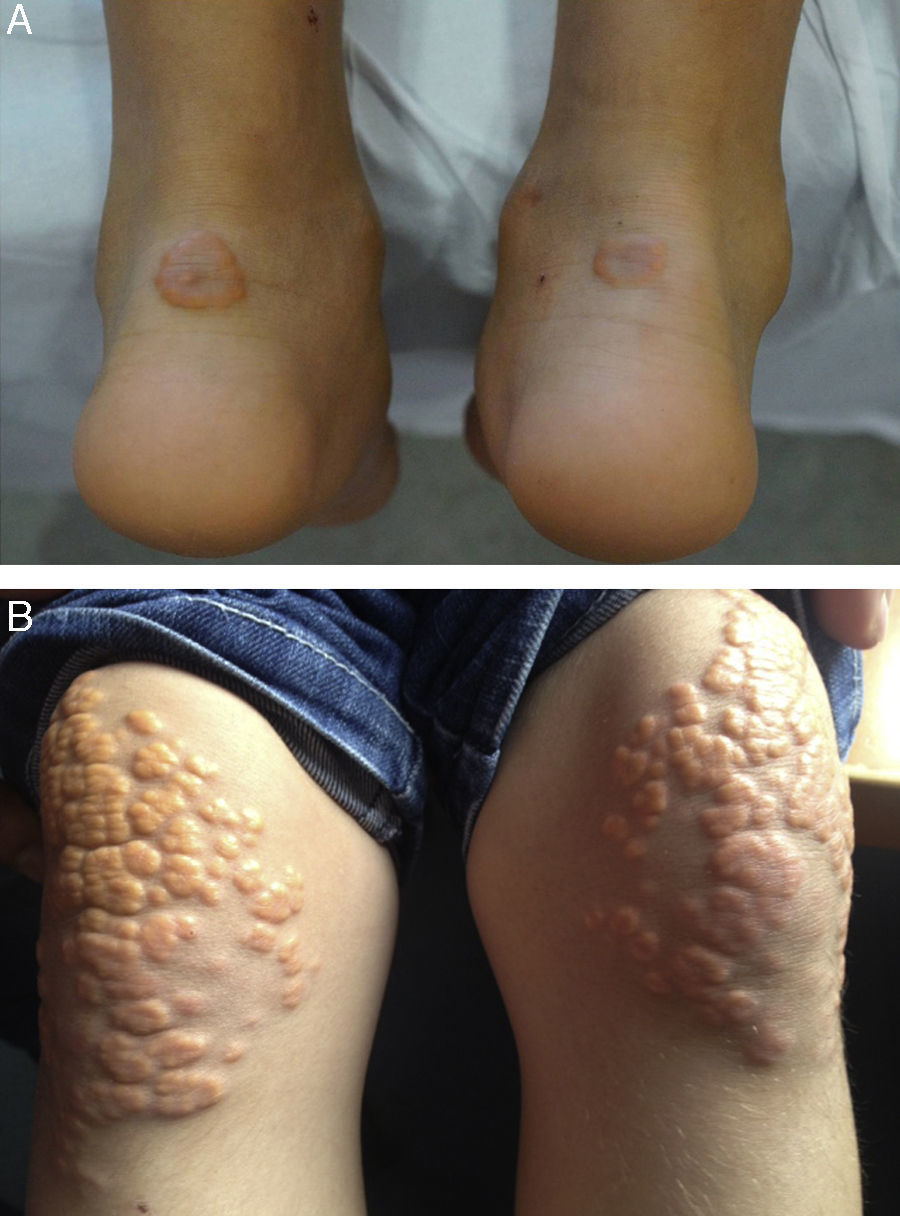
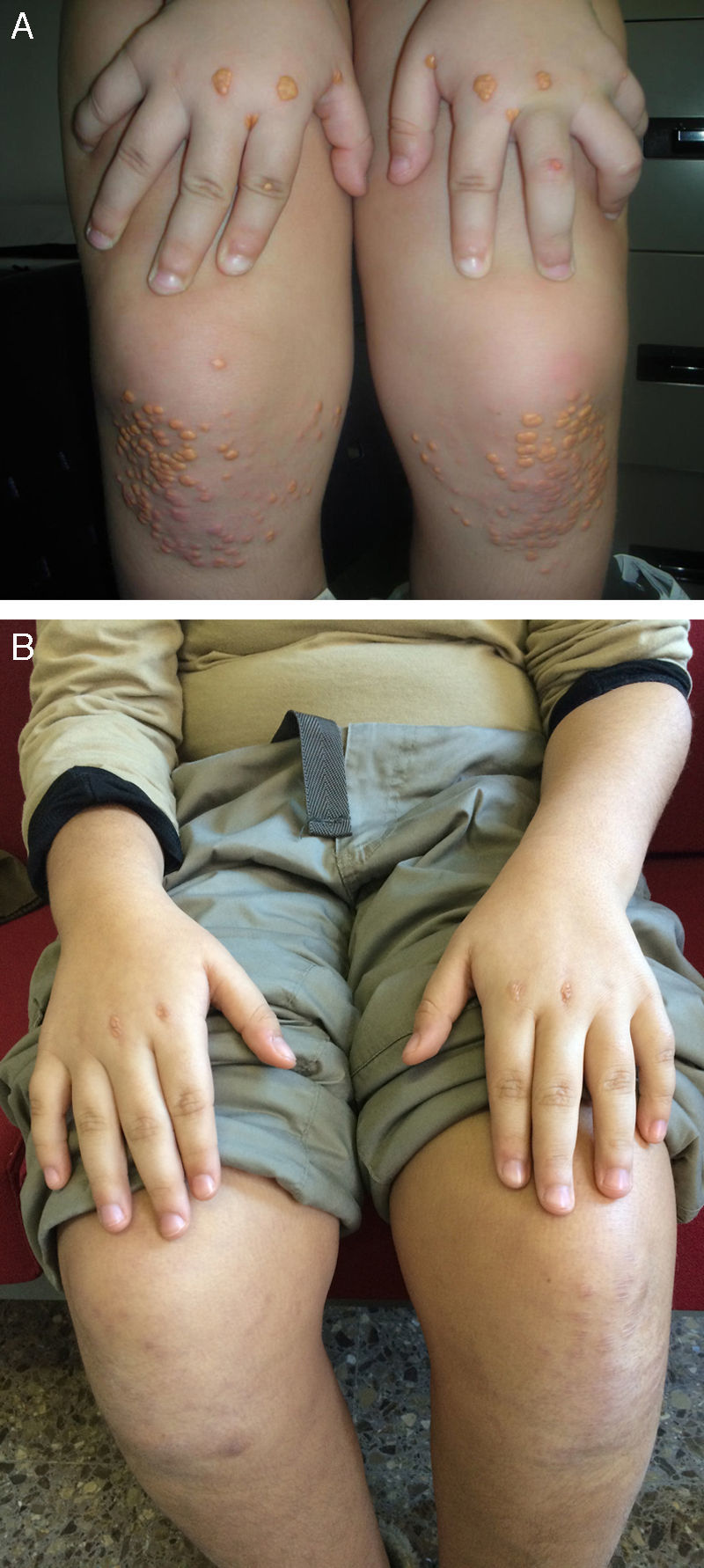
Xantomas y arco cornealAunque no se asocia exclusivamente con la HFHo, la presencia de xantomas cutáneos y/o tendinosos en niños es altamente sugestiva del diagnóstico (fig. 5). La presencia de arco corneal refuerza el diagnóstico clínico. Como se observa para los niveles de cLDL, la variabilidad en la edad de aparición y en la extensión de los xantomas se puede explicar parcialmente por las mutaciones subyacentes, con una aparición más temprana de xantomas asociada al estado de receptor negativo frente a receptor defectuoso2,13. Los depósitos de colesterol en los tendones y articulaciones pueden causar tendinitis y dolor articular, lo que afecta a la calidad de vida de los pacientes y puede requerir su extirpación quirúrgica. En casos excepcionales, los pacientes pueden presentar xantomas ectópicos gigantes en el cerebro, mediastino y músculos de los glúteos20. La aparición de xantomas en niños pequeños es con frecuencia el factor clave del diagnóstico de la HFHo2,21-23, por lo que su reconocimiento inmediato es crucial para un diagnóstico precoz. La presencia de niveles marcadamente elevados de cLDL y la ausencia de síntomas neurológicos, cognitivos y oftálmicos en pacientes con HFHo los distingue de los pacientes con xantomatosis cerebrotendinosa24.
Antecedentes familiares y el Dr. Pedro Mata (B).")
Es esencial una evaluación cuidadosa de los antecedentes familiares para la valoración exhaustiva de una posible HF en general y de una HFHo en particular8. En el caso de las mutaciones autosómicas dominantes (en genes LDLR, PCSK9 y APOB), ambos progenitores son heterocigotos obligados y, por tanto, muestran niveles elevados de cLDL (frecuentemente percentil >95 según los criterios de edad y sexo específicos del país) y antecedentes familiares positivos de ECVA prematura (<55años en varones y <60años en mujeres entre familiares de primer grado). En el caso de la hipercolesterolemia autosómica recesiva (debida a mutaciones en LDLRAP1), los progenitores pueden presentar niveles de cLDL en el intervalo normal, y la determinación de un árbol genealógico extenso de la familia puede revelar un patrón autosómico recesivo heredado. El cribado en cascada familiar ofrece prospectivamente a los padres con HFHe la posibilidad de consejo genético e identificar a pacientes con HFHo al nacer, permitiendo así el inicio precoz del tratamiento. La identificación de la HFHo también puede guiar la detección en cascada «inversa» para progenitores y familiares a fin de identificar a los pacientes con HF.
Diferenciación de la sitosterolemiaAunque en la mayoría de los casos el diagnóstico de HFHo es relativamente sencillo, otro trastorno del metabolismo lipídico, la sitosterolemia (también llamada «fitosterolemia»), puede tener una presentación clínica similar, con la presencia de xantomas tendinosos y/o tuberosos en niños asociada a un notable aumento de los niveles de colesterol en plasma y complicaciones arterioscleróticas25. Sin embargo, es relevante que la enfermedad arteriosclerótica no siempre esté presente en los sujetos sitosterolémicos definidos genéticamente, como se muestra en una reciente publicación26. Al igual que la hipercolesterolemia autosómica recesiva, la sitosterolemia tiene un patrón de herencia autosómica recesiva, por lo que los progenitores pueden presentar niveles normales de colesterol. Dos características importantes diferencian la sitosterolemia de la HFHo: a)concentraciones plasmáticas marcadamente elevadas (>30veces) de fitoesteroles25, y b)niveles elevados de colesterol que responden bien a la dieta y a los secuestrantes de ácidos biliares o ezetimiba y que pueden desaparecer después de las 2 primeras décadas de vida25,26. El diagnóstico se confirma mediante análisis genético, con mutaciones en 2 genes de los ATP binding cassette transporters, ABCG5 y/o ABCG8, que son los causantes de la sitosterolemia25.
En resumen, este Grupo de Consenso recomienda que el diagnóstico se realice mediante una cuidadosa evaluación de las características clínicas y de los antecedentes familiares, así como mediante pruebas genéticas, especialmente cuando el diagnóstico clínico de la HFHo sea dudoso o para facilitar el cribado en cascada familiar «inversa». La detección en cascada familiar «inversa» se recomienda siempre en cualquier caso.
Complicaciones cardiovasculares y evolución naturalLa presencia de niveles marcadamente elevados de cLDL en plasma a partir del nacimiento tiene como secuela la aparición de complicaciones de la ECVA únicas en la HFHo8. El valor de colesterol-año, una medida que integra la gravedad y duración de la hipercolesterolemia, está directamente asociado a la presencia de depósitos de colesterol en compartimentos vasculares y extravasculares en pacientes con HFHo27, lo que refuerza el concepto de que los niveles absolutos de cLDL afectan a la gravedad del fenotipo de manifestaciones cardiovasculares (CV). En pacientes con HFHo diagnosticada clínicamente se producen a menudo episodios CV durante la adolescencia2,28,29, y se han descrito angina de pecho, infarto de miocardio y muerte en la primera infancia, habitualmente en individuos con LDLR negativo1,2,21-23,28. Los pacientes con HFHo no tratados que son LDLR negativo raramente sobreviven más allá de su segunda década de vida. Aunque los pacientes con HFHo con LDLR defectuoso tienen mejor pronóstico, casi todos desarrollan ECVA clínicamente significativa hacia los 30años de edad. Sigue siendo necesario realizar estudios a largo plazo para evaluar el riesgo CV en la HFHo confirmada genéticamente sin el fenotipo grave que se observa habitualmente en la HFHo clínicamente definida.
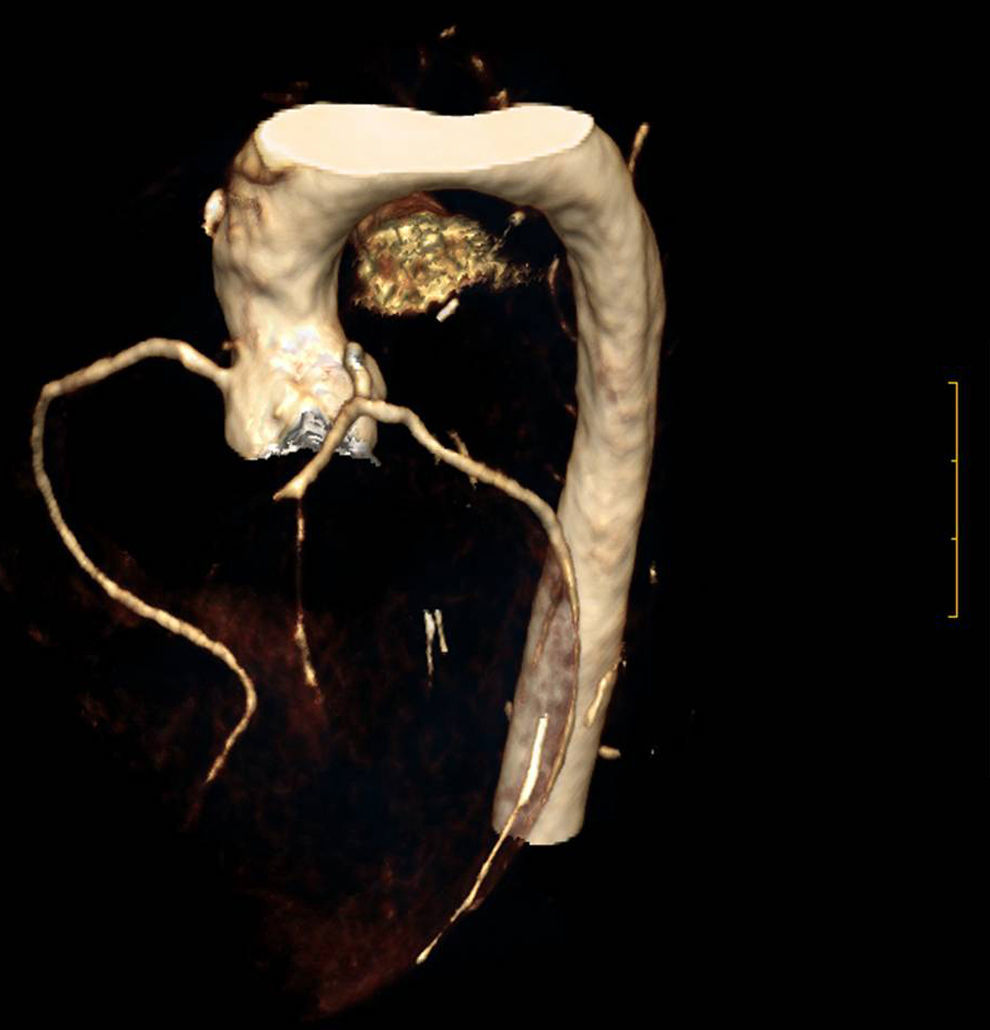
La HFHo se caracteriza por una arteriosclerosis acelerada que afecta la raíz aórtica y daña el ostium coronario y las arterias coronarias, pudiendo afectar a otras áreas, como la carótida, la aorta descendente y las arterias ileofemoral y renal1,30. Los depósitos de colesterol y calcio, así como la fibrosis y la inflamación, tanto en la raíz aórtica como en las valvas de la válvula aórtica, pueden causar estenosis aórtica a nivel valvular y supravalvular (fig. 6)1,31. Estas manifestaciones se producen frecuentemente en la primera y segunda década de vida1,2,28,30. Los pacientes pueden permanecer inicialmente asintomáticos, presentando únicamente xantomas cutáneos y tendinosos y un soplo cardiaco en el área aórtica2,32. Con frecuencia se observa afectación temprana de la aorta torácica ascendente y descendente1, acompañada de calcificación aórtica grave y prematura en pacientes adultos33. También se ha descrito lesión de la válvula mitral por depósito de colesterol en las valvas1.

Es de destacar que la enfermedad aórtica, valvular y supravalvular, puede progresar, incluso cuando los niveles de colesterol han sido reducidos, como consecuencia del estrés hemodinámico y la fibrosis progresiva en las regiones afectadas31. También son frecuentes la disnea, la insuficiencia cardiaca izquierda y la muerte súbita cardiaca1,30. En niños pequeños, los primeros síntomas y signos suelen estar ligados a la regurgitación y la estenosis aórtica2. La angina de pecho, consecuencia tanto de la reducción en el suministro de oxígeno causada por la arteriosclerosis coronaria como del aumento de la demanda del ventrículo izquierdo (debido a su hipertrofia y a la obstrucción del flujo de salida), pueden producirse a cualquier edad, dependiendo de la velocidad de progresión y de la gravedad del fenotipo (tabla 2).
Complicaciones cardiovasculares de la hipercolesterolemia familiar homocigota
| • La HFHo se caracteriza por una arteriosclerosis acelerada que normalmente afecta a la raíz aórtica, aunque también pueden estar afectadas otras regiones vasculares |
| • Los primeros episodios cardiovasculares importantes a menudo se producen durante la adolescencia, e incluso a edades más tempranas cuando los pacientes son LDLR negativo o no han recibido tratamiento |
| • En niños pequeños, los primeros síntomas y signos están a menudo ligados a la regurgitación y la estenosis aórtica, debido a la acumulación de colesterol a nivel valvular |
| • Puesto que la afectación valvular y supravalvular aórtica puede progresar incluso cuando los niveles de colesterol han sido reducidos, está indicada la evaluación periódica de la cardiopatía aórtica, carotídea y coronaria subclínicas |
Considerando el riesgo extremadamente alto de aparición temprana de ECVA grave y su rápida progresión en la HFHo, está indicado el cribado regular para cardiopatía aórtica y coronaria subclínicas. Este Grupo de Consenso recomienda que los pacientes se sometan a una evaluación CV completa durante el diagnóstico, con la posterior evaluación anual mediante ecografía doppler de corazón y aorta y, si está disponible, angio-TAC cada 5años, o con más frecuencia si está clínicamente indicado, teniendo en cuenta la exposición a la radiación y la gravedad de la enfermedad subclínica. Siempre que la edad del paciente lo permita y se pueda otorgar el debido consentimiento, se puede realizar una angio-TAC coronaria que permite detectar la obstrucción de la luz del vaso por placas calcificadas y no calcificadas34; además, sus resultados se pueden combinar con los de las pruebas de esfuerzo miocárdico. Las pruebas de esfuerzo, aunque no son óptimas para detectar la enfermedad subclínica, se pueden emplear en caso de acceso limitado a la angio-TAC coronaria o a la resonancia magnética (RM) cardiaca. Debido a la preocupación que suscita la exposición de individuos jóvenes a la radiación, la angio-TAC coronaria debe realizarse en escáneres de TC con al menos 64 y preferiblemente 320 detectores o escáneres de fuente doble, con la exposición a la radiación adaptada al peso corporal35. El grado de afectación arteriosclerótica de la aorta también puede evaluarse mediante RM36 o ecocardiograma transesofágico37.
Las pruebas de esfuerzo y la angiografía coronaria invasiva están indicadas en pacientes con síntomas clínicos indicativos de isquemia o mal funcionamiento valvular, o si la evaluación cardiaca no invasiva ha dado resultados positivos. Dada la alta tasa de estenosis del ostium, el riesgo de muerte súbita y la incapacidad de realizar las pruebas de esfuerzo debido a la edad, en niños pequeños gravemente afectados puede estar indicada la angiografía invasiva. Esta intervención debe ser realizada por un cardiólogo intervencionista pediátrico con experiencia. La revascularización coronaria está indicada en el caso de cardiopatía coronaria grave, y la sustitución de la válvula aórtica, en el caso de obstrucción grave del flujo de salida del ventrículo izquierdo. Con cualquier tipo de intervención quirúrgica debe tenerse cuidado con la raíz aórtica, ya que habitualmente está gravemente afectada por placas arterioscleróticas y calcificaciones. La reconstrucción de la raíz aórtica podría ser necesaria con la sustitución de la válvula aórtica38. El seguimiento de estos pacientes debe ser realizado por un equipo de expertos formado por cardiólogos y especialistas en lípidos que trabajen en estrecha colaboración para optimizar la medidas terapéuticas, lo que incluye el tratamiento farmacológico antiplaquetario, la prevención de la endocarditis, especialmente en aquellos pacientes con prótesis valvular o injertos aórticos, e intervenciones quirúrgicas destinadas a corregir la insuficiencia valvular y coronaria.
Tratamiento actual para la hipercolesterolemia familiar homocigotaDadas las graves complicaciones de la ECVA asociadas a la HFHo, es fundamental reducir la carga que suponen los niveles elevados de cLDL. Es preciso fomentar una dieta cardiosaludable baja en grasas saturadas y en colesterol en todos los pacientes con HFHo, aproximándose lo más posible al patrón nutricional de la dieta mediterránea, aunque, incluso con una adherencia estricta, la dieta tiene poco impacto sobre la gravedad de la hipercolesterolemia. Es necesario animar a los pacientes a llevar una vida activa. Puesto que la estenosis aórtica puede precipitar la angina y el síncope durante el ejercicio, se recomienda una evaluación cuidadosa de la afectación aórtica y del ostium antes de iniciar una actividad deportiva. Otros factores de riesgo, como el tabaquismo, la hipertensión y la diabetes, aunque menos frecuentes que en la edad adulta, deben tratarse de forma agresiva si están presentes.
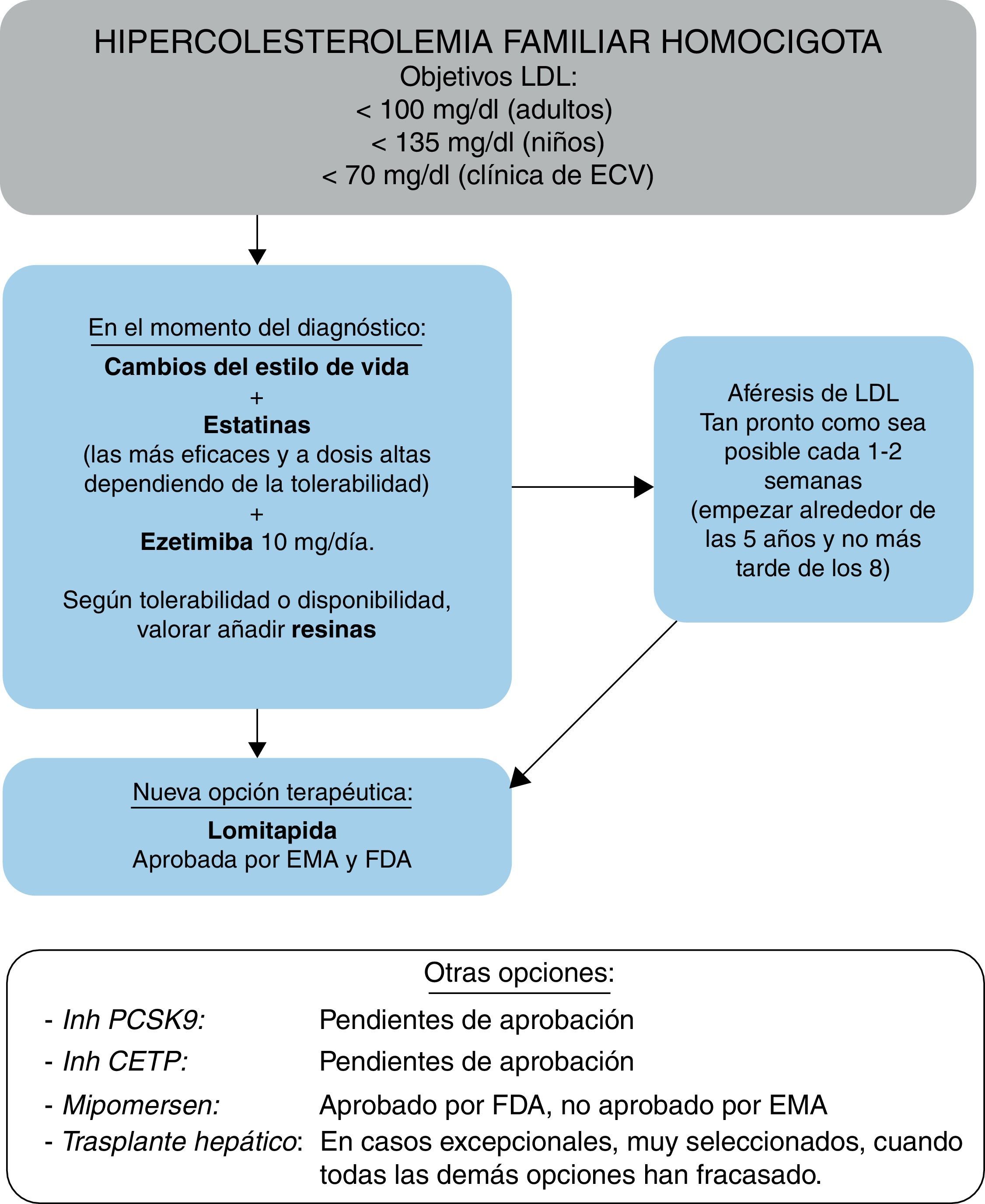
FarmacoterapiaEste Grupo de Consenso recomienda que la terapia hipolipemiante se inicie lo antes posible, teniendo en cuenta las evidencias de que el tratamiento puede retrasar la aparición de episodios clínicos de ECVA2,28. De acuerdo con las guías recientemente publicadas8, los objetivos terapéuticos del nivel de cLDL en pacientes con HFHo son <100mg/dl (<2,5mmol/l) en adultos; <135mg/dl (<3,5mmol/l) en niños, o <70mg/dl (<1,8mmol/l) en pacientes con ECVA clínica. Sin embargo, este Grupo de Consenso es consciente de que estos objetivos son de difícil consecución y reconoce que la heterogeneidad genética y fenotípica de la HFHo puede traducirse en una amplia variabilidad de la respuesta a las terapias hipolipemiantes, tanto convencionales como novedosas.
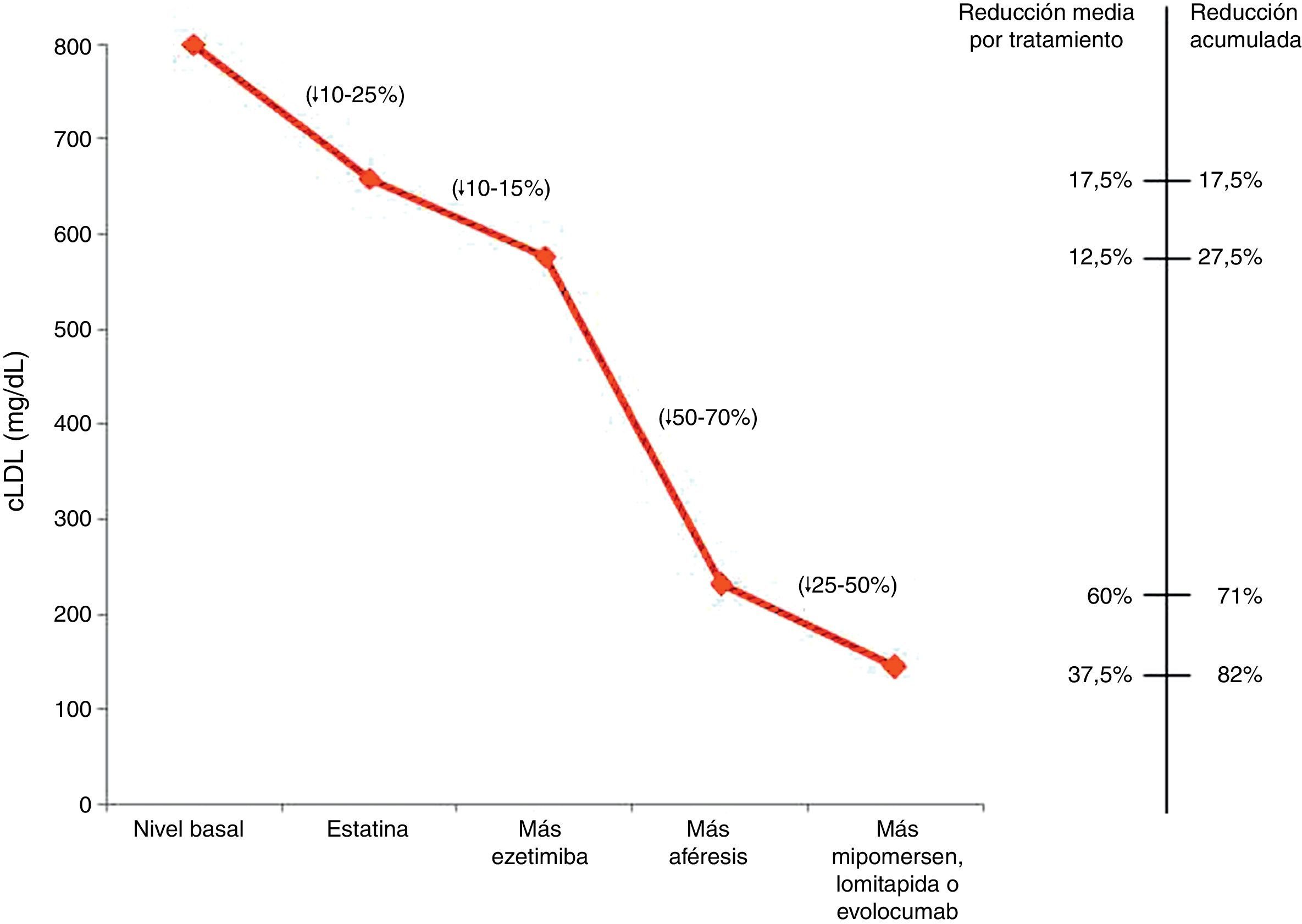
Está comprobada la eficacia de las estatinas como pilar del tratamiento en la HFHo, incluso en individuos con receptor negativo39-43, habiéndose demostrado que reducen la mortalidad CV y por todas las causas28. No obstante, incluso a las dosis más altas de las estatinas más eficaces (atorvastatina y rosuvastatina), en la mayoría de los pacientes solo se observan reducciones modestas de los niveles plasmáticos de cLDL, de entre el 10 y el 25 %. La hipercolesterolemia autosómica recesiva parece que responde relativamente mejor al tratamiento44. La adición del inhibidor de la absorción del colesterol, ezetimiba, produce una reducción de los niveles de cLDL de un 10-15%45, por lo que su adición a estatinas proporciona una reducción del 30-40% en los niveles de cLDL, con efectos adversos mínimos y costes relativamente bajos (fig. 7), habiéndose demostrado recientemente su impacto beneficioso en la reducción de episodios CV46. Las combinaciones de estatinas y ezetimiba con otros medicamentos hipolipemiantes, como secuestrantes de ácidos biliares y fibratos, se pueden considerar, en tercera línea, para incrementar la reducción de cLDL, aunque su uso puede estar limitado por su tolerabilidad y disponibilidad. En nuestro país, el Sistema Nacional de Salud otorgó la aportación reducida al tratamiento crónico con estatinas, ezetimiba y resinas (colesevelam).

Efecto acumulado sobre la reducción del cLDL en la HFHo mediante la utilización de estatina, ezetimiba, aféresis de LDL y mipomersen, lomitapida o evolocumab. El porcentaje de reducción del cLDL depende de los valores basales de cLDL. En la figura se muestra la disminución del cLDL tras un único tratamiento de aféresis de LDL. Tras el tratamiento, el nivel de cLDL es mayor en los pacientes que presentaban valores basales más altos. Sin embargo, las curvas de rebote tras el tratamiento son más o menos paralelas. Véase Schuff-Werner et al.51.
La eliminación extracorpórea de cLDL, aunque sea una estrategia terapéutica cara y más o menos compleja, es coste-efectiva y debe considerarse como un tratamiento eficaz para la HFHo47-50, debiendo remitirse al paciente a un centro especializado en aféresis de LDL. El enfoque inicial no selectivo de recambio plasmático se ha sustituido por varios métodos de eliminación selectiva de lipoproteínas aterogénicas51, que muestran una eficacia terapéutica similar. Un tratamiento puede reducir los niveles de cLDL en plasma en un 55-70 % respecto a los niveles previos al tratamiento, pudiéndose alcanzar niveles de cLDL próximos a los normales cuando la aféresis de LDL se realiza semanalmente. El tratamiento prolongado a menudo logra la regresión de las lesiones vasculares y los xantomas cutáneos (fig. 8). A pesar de la falta de estudios aleatorizados, existen evidencias clínicas que indican que el uso prolongado de la aféresis de LDL puede contribuir a la regresión de la placa y/o a la estabilización y mejora del pronóstico, como se ha revisado de forma exhaustiva51, y es rentable en la HFHo, especialmente en fenotipos graves (véase en Internet el material complementario del consenso europeo: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4139706/bin/supp_35_32_2146__index.html). Queda por establecer si otros efectos, como la importante reducción en Lp(a)50, la modificación de los fenómenos inflamatorios vasculares, la mejora en las condiciones reológicas y los cambios en la expresión de genes en las células sanguíneas, añaden beneficios al pronóstico CV. Los datos acumulados son fuertemente indicativos de que cuanto antes se inicia la aféresis de LDL, mejor es el pronóstico. En la práctica, la edad de inicio y la frecuencia del tratamiento representan un compromiso entre la viabilidad práctica, el coste y la necesidad clínica de lograr el objetivo de cLDL. A pesar de que la frecuencia teórica óptima es un procedimiento a la semana, la mayoría de los centros tratan a los pacientes cada 2semanas. Debe continuarse con la aféresis de LDL durante el embarazo en este grupo de pacientes, ya que no está contraindicada48,50.
 y a la edad de 11años tras 4años de tratamiento con aféresis de LDL (B).Las fotografías han sido amablemente cedidas por el Dr. Pedro Mata (A) y la Dra. Cristina Arbona (B).")
La aféresis de LDL es bien tolerada, habiéndose descrito efectos adversos en menos del 5% de los procedimientos52, que incluyen hipotensión, dolor abdominal, náuseas, hipocalcemia y anemia por deficiencia de hierro. El uso concomitante con inhibidores de la enzima convertidora de angiotensina puede condicionar hipotensión grave con algunos de los métodos selectivos (celulosa dextrano, hemoperfusión e inmunoadsorción), por lo que deben evitarse o suspenderse al menos 24h antes del procedimiento, pudiendo reiniciarlo al terminar la sesión53. En pacientes muy jóvenes54,55 el acceso venoso suele un problema, siendo necesario la colocación de un catéter venoso central; por ello, a las complicaciones del procedimiento debemos añadir sus posibles efectos secundarios, infección y trombosis52.
En línea con las guías actuales47-49, este Grupo de Consenso recomienda que se considere la aféresis de LDL en pacientes con HFHo. El tratamiento debe iniciarse lo antes posible, preferiblemente a los 5años y no más tarde de los 8, aunque esto y la frecuencia de tratamiento representan un compromiso entre el acceso a los centros, la gravedad de la enfermedad, el coste y la elección del paciente56-58. Esta opción terapéutica está dentro de la cartera de servicios del Ministerio de Sanidad y se debe considerar para los pacientes con HFHo.
Trasplante hepático y otros enfoques quirúrgicosEs una opción prácticamente en desuso en la actualidad. El trasplante hepático corrige el defecto molecular en el órgano más activo en el aclaramiento de LDL, lo que produce una notable mejora de los niveles de cLDL1, con reducciones hasta del 80%59-61. Aunque se trata de una estrategia terapéutica eficaz, tanto por sí sola como en combinación con un trasplante de corazón62-64, el porcentaje de reducción de cLDL que se alcanza con el mismo se puede conseguir en la actualidad con la combinación de estilo de vida, fármacos y aféresis de LDL (fig. 8). Por otra parte, presenta desventajas obvias, como el alto riesgo de complicaciones quirúrgicas, la mortalidad postrasplante, la escasez de donantes y la necesidad de tratamiento de por vida con inmunosupresores65. Por todo ello, consideramos el trasplante hepático como una medida excepcional que solo debe plantearse cuando se han agotado el resto de las medidas terapéuticas. La derivación porto-cava y el by-pass ileal son técnicas de carácter histórico, sin fundamento en la actualidad.
En conclusión, este Grupo de Consenso hace hincapié en la necesidad de una combinación de estilo de vida, tratamiento con estatinas y ezetimiba (con o sin otros fármacos hipolipemiantes) y aféresis de LDL para tratar la HFHo. Aunque este enfoque puede ser suficiente para conseguir el objetivo de cLDL en pacientes con un fenotipo «más leve», el Grupo reconoce que la HFHo es normalmente resistente a los tratamientos hipolipemiantes existentes y que la aféresis de LDL presenta limitaciones prácticas. Nuevos fármacos hipolipemiantes con diferentes mecanismos de acción podrían mejorar el tratamiento de esta afección (fig. 9).
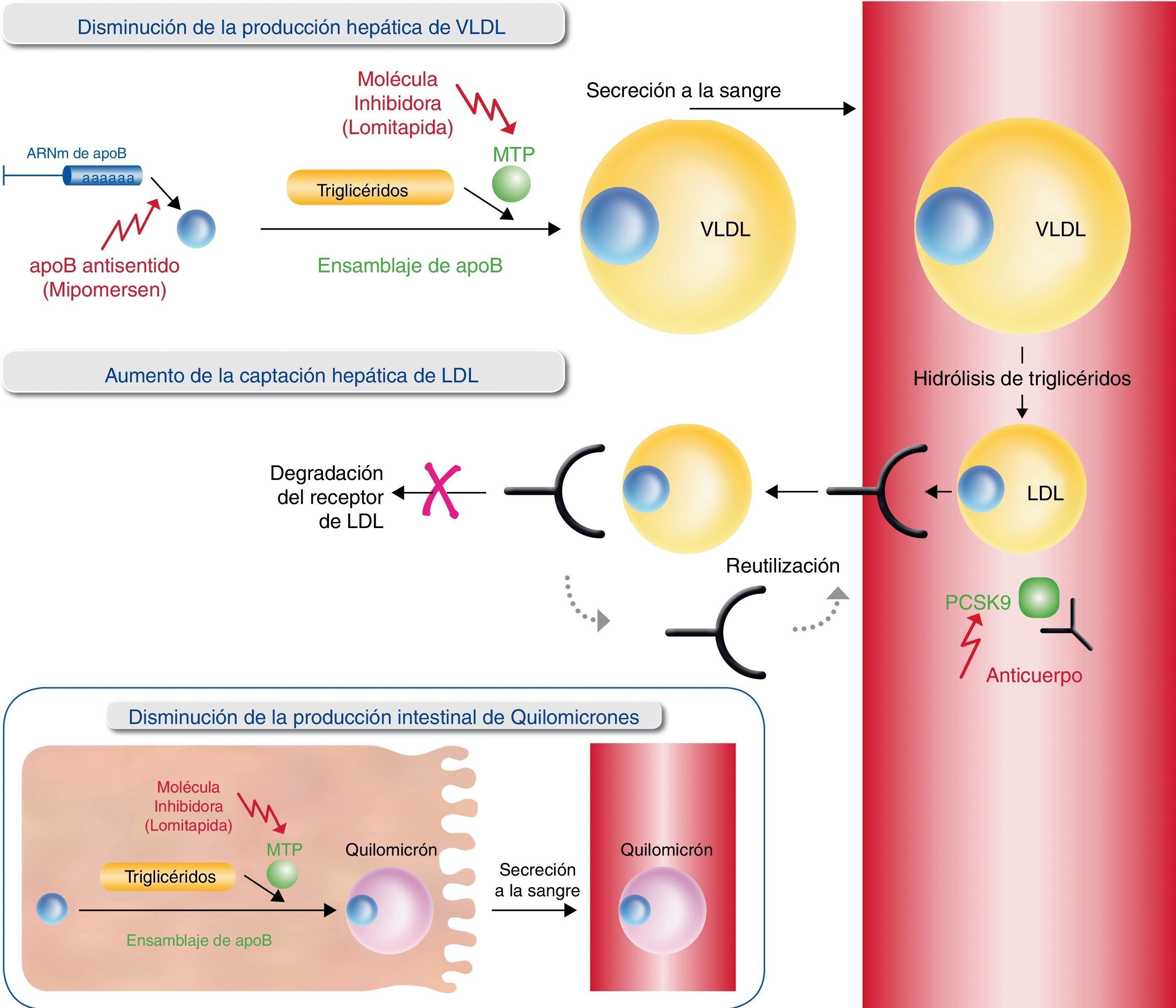
![Dianas de los nuevos fármacos reguladores de lípidos. Los nuevos fármacos pretenden influir en la producción de lipoproteínas de muy baja densidad (VLDL), inhibiendo la síntesis de apolipoproteína B (oligonucleótido antisentido de la apolipoproteína B [apoB], mipomersen), la carga de lípidos en apoB naciente (inhibidor de la proteína de transferencia microsómica de triglicéridos [MTP], lomitapida) o en el catabolismo de LDL aumentando el reciclaje del receptor de LDL (inhibidores de PCSK9).](https://static.elsevier.es/multimedia/02149168/0000002700000002/v2_201503280426/S0214916815000042/v2_201503280426/es/main.assets/gr9.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcxT+B4FjbiPdoCJto3d5S/Ch6vvG+Cp80/Jya7nfY0UrJ5X6BLWiNhEjkiUXVdtWXJK/gEPXW+JvLnq83CriJS7copuCeAaH9/oHh0uyiKfgiIUjS6oVYDm1lMLeAbibEhaX+lcbxlh7Ey8zWWJ7bGQRHCd3p5h4nG8F2EhvPqHPXs0ny6Uvk0afs+JjUabraGb4oyRmlH+7BzflADlKXFNzgOBS1bPKSwNGRNFxUUIZt3KIEepfzWYN4clzkis18= "Dianas de los nuevos fármacos reguladores de lípidos. Los nuevos fármacos pretenden influir en la producción de lipoproteínas de muy baja densidad (VLDL), inhibiendo la síntesis de apolipoproteína B (oligonucleótido antisentido de la apolipoproteína B [apoB], mipomersen), la carga de lípidos en apoB naciente (inhibidor de la proteína de transferencia microsómica de triglicéridos [MTP], lomitapida) o en el catabolismo de LDL aumentando el reciclaje del receptor de LDL (inhibidores de PCSK9).")
Dianas de los nuevos fármacos reguladores de lípidos. Los nuevos fármacos pretenden influir en la producción de lipoproteínas de muy baja densidad (VLDL), inhibiendo la síntesis de apolipoproteína B (oligonucleótido antisentido de la apolipoproteína B [apoB], mipomersen), la carga de lípidos en apoB naciente (inhibidor de la proteína de transferencia microsómica de triglicéridos [MTP], lomitapida) o en el catabolismo de LDL aumentando el reciclaje del receptor de LDL (inhibidores de PCSK9).
Recientemente han surgido nuevas moléculas cuyo mecanismo de acción se basa en alterar la producción y secreción de lipoproteínas con apoB66,67, en lugar de aumentar su eliminación de la circulación, capacidad esta última gravemente alterada en pacientes con HFHo. Es por ello que este tipo de enfoque resulta prometedor para el tratamiento de la hipercolesterolemia. Estos fármacos son lomitapida, recientemente aprobada por la Food and Drug Administration (FDA) y por la Agencia Europea del Medicamento (EMA) como terapia complementaria en pacientes mayores de 18años con o sin aféresis de LDL, y mipomersen, aprobado por la FDA y rechazado por la EMA, también como terapia complementaria en pacientes mayores de 12años sin aféresis de LDL.
LomitapidaLomitapida es un inhibidor oral de la proteína de transferencia microsómica de triglicéridos (MTP), que es responsable de transferir triglicéridos y fosfolípidos a quilomicrones y VLDL durante su ensamblaje en el intestino y el hígado, respectivamente68. La inhibición de MTP induce una reducción de la secreción de estas lipoproteínas a la circulación. En un ensayo clínico abierto en pacientes con HFHo, lomitapida a las dosis máximas toleradas, junto con el tratamiento estándar que podía incluir aféresis de LDL, redujo los niveles plasmáticos de cLDL y apoB ∼50% y de Lp(a) ∼15% a las 26semanas, con una reducción duradera de cLDL durante un periodo adicional de seguimiento de 12meses19. Los acontecimientos adversos observados con mayor frecuencia fueron síntomas gastrointestinales y acumulación de grasa hepática. Los acontecimientos adversos gastrointestinales (p.ej., náuseas, flatulencia y diarrea) se redujeron mediante un régimen de aumento progresivo de la dosis combinado con el seguimiento de una dieta baja en grasas (<20% del aporte de calorías proveniente de las grasas) y separando de las comidas la administración del fármaco19. Se observaron elevaciones de los niveles de alanina aminotransferasa (ALT) superiores a 3veces el límite superior de la normalidad (LSN) en el 34% de los pacientes19. Se ha observado una acumulación de grasa hepática hasta una mediana del 9% (intervalo de 0-34%) a las 26semanas y del 8% (0-19%) a las 78semanas19. En España se ha publicado la primera experiencia clínica con dicho fármaco69.
MipomersenMipomersen, no aprobado en Europa, es un oligonucleótido antisentido, administrado mediante inyección subcutánea, que tiene como objetivo el ácido ribonucleico mensajero (ARNm) de la apoB66. En un ensayo clínico doble ciego controlado con placebo en pacientes con HFHo, mipomersen junto con el tratamiento hipolipemiante de referencia tenía como resultado reducciones de los niveles plasmáticos de cLDL a las 26semanas (media del 25%), apoB (27%) y Lp(a) (31%) en comparación con placebo70. Los acontecimientos adversos notificados con mayor frecuencia fueron reacciones en el lugar de inyección (76% de los pacientes), algunas de las cuales fueron de larga duración, y síntomas pseudogripales que aparecieron 2días después de la inyección70. Se han notificado elevaciones en los niveles de ALT durante el tratamiento con mipomersen; en el 12% de los pacientes se observaron aumentos superiores a 3veces el LSN70. El contenido de grasa hepática no se midió de forma rutinaria en el estudio de HFHo con mipomersen, aunque en pacientes con hipercolesterolemia, incluidos aquellos con HFHe, se observó una mediana de aumento de ∼5% (intervalo de −1 a 37%) después de 28semanas de tratamiento71.
Limitaciones de la terapiaEl aumento del contenido en grasa hepática observado durante el tratamiento tanto con lomitapida como con mipomersen puede correlacionarse con el grado de eficacia19,70. Los limitados datos disponibles sugieren que este efecto es reversible tras la suspensión del tratamiento. Debido al posible riesgo de hepatotoxicidad, ambos fármacos han sido aprobados por la FDA para un uso restringido. Aunque los posibles beneficios CV asociados a una disminución sustancial de cLDL posiblemente superen el teórico aumento del riesgo de esteatohepatitis y fibrosis en dichos pacientes de muy alto riesgo, se necesita una evaluación sistemática de la eficacia, del resultado y de la seguridad hepática a largo plazo. Además, ambos fármacos presentan acontecimientos adversos que pueden limitar su uso prolongado. Lomitapida está también contraindicada con inhibidores potentes y moderados de CYP3A4.
Indudablemente, las estatinas y otros fármacos hipolipemiantes convencionales están disponibles en todo el mundo y son asequibles, en comparación con la aféresis de LDL y las opciones de tratamiento más recientes. Aunque el elevado coste de estos enfoques terapéuticos deba ser tenido en cuenta, el importe total de tratar adecuadamente la HFHo sigue siendo bajo debido a la rareza de la enfermedad y al hecho de descontar el coste de los tratamientos asociados a las complicaciones CV.
Opciones futurasHay varios fármacos nuevos que también pueden ofrecer opciones terapéuticas para la HFHo. Se están desarrollando tratamientos con anticuerpos monoclonales dirigidos frente a PCSK9 (fig. 9). En pacientes con HFHe, este tratamiento redujo los niveles de cLDL hasta en un 65% en combinación con las terapias hipolipemiantes concomitantes72,73. Posteriormente, un estudio preliminar mostró que evolocumab (AMG 145, 420mg por vía subcutánea cada 2semanas) reducía los niveles plasmáticos de cLDL en un 26% (media de 116mg/dl o 3mmol/l) en pacientes con HFHo que tenían receptores defectuosos74; en un ensayo clínico en fase3 controlado con placebo, evolocumab redujo los niveles de cLDL en −23% vs +8% placebo (diferencia: 31%)75. Sin embargo, evolocumab fue muy poco eficaz en los pacientes que presentan HFHo con receptores negativos. Puesto que los niveles notablemente elevados de PCSK9 se asocian con la HFHo, tanto con tratamiento como sin tratamiento con estatinas76, el uso de inhibidores de PCSK9 como terapia complementaria en sujetos con mutaciones de LDLR defectuoso o mutaciones de ganancia de función de PCSK9 ofrece un enfoque terapéutico adicional para optimizar la reducción de cLDL.
Otros temasAdemás del tratamiento farmacológico de la hipercolesterolemia, el tratamiento del paciente con HFHo debe abordar otros aspectos (tabla 3). El diagnóstico de la HFHe puede afectar al funcionamiento psicosocial y a la calidad de vida, incluso más intensamente en el caso de los pacientes con HFHo, lo que supone la necesidad de integrar el apoyo psicológico dentro de la atención médica habitual del paciente77. Por tanto, este Grupo recomienda que se informe adecuadamente a padres e hijos sobre la HFHo, proporcionando de este modo una base para la toma de decisiones compartidas respecto a su tratamiento.
Resumen de las recomendaciones del Grupo de Consenso de la SEA y la FHF
| • DIAGNÓSTICO:- Criterios de diagnóstico:• Confirmación genética de dos alelos portadores de mutaciones funcionales en el locus del gen LDLR, APOB, PCSK9 o LDLRAP1 OcLDL sin tratamiento ≥500 mg/dL (≥13 mmol/L) o cLDL con tratamiento ≥300 mg/dL (≥8 mmol/L) (estos niveles de cLDL son solo indicativos, y niveles más bajos, especialmente en niños o en pacientes tratados, no excluyen HFHo), junto con:- Xantomas cutáneos y/o tendinosos evidenciados antes de los 10 años de edad O- Niveles elevados de cLDL sin tratamiento en ambos progenitores, compatible con HFHe- Los pacientes en los que se sospeche el diagnóstico, deberán remitirse a centros especializados para un tratamiento completo adecuado.- Debe considerarse siempre un análisis genético y especialmente para:• Confirmar el diagnóstico clínico• Facilitar las pruebas de los miembros de la familia• Ayudar en el diagnóstico en casos dudosos• CRIBADO PARA ECVA SUBCLÍNICA- Los pacientes deberían someterse a una evaluación CV completa durante el diagnóstico, con la posterior evaluación anual mediante ecografía doppler de corazón y aorta. Otras pruebas de imagen dependiendo del contexto clínico.• TRATAMIENTO- Se basa en una combinación de cambios del estilo de vida, estatinas con ezetimiba y aféresis de LDL.- La terapia hipolipemiante con fármacos debe iniciarse lo antes posible (a partir de los 2 años).- Se considerará la aféresis de LDL en todos los pacientes con HFHo y se iniciaría lo antes posible, preferiblemente a los 5 años y no más tarde de los 8 años.- Lomitapida debería considerarse como tratamiento complementario para reducir adicionalmente los niveles plasmáticos de cLDL en pacientes con HFHo, con o sin aféresis.• OTROS TEMAS- Los métodos hormonales anticonceptivos están contraindicados en la HFHo. Las mujeres que deseen quedarse embarazadas deberán recibir asesoramiento y someterse a una evaluación CV. Las mujeres embarazadas deberán tratarse con aféresis de LDL.- El apoyo psicológico debería integrarse dentro de la atención médica habitual.- Los grupos de apoyo al paciente y a la familia tienen un papel relevante.- Puede considerarse la cirugía para extirpar xantomas cutáneos y/o tendinosos grandes por motivos funcionales o estéticos. |
Los métodos anticonceptivos y el embarazo deberán discutirse cuidadosamente con las pacientes; los métodos hormonales están contraindicados en la HFHo, por lo que se recomienda claramente el uso de otros métodos anticonceptivos (tabla 3). También merecen consideración las consecuencias del embarazo, es decir, el agravamiento de la hipercolesterolemia por causa de la interrupción de la farmacoterapia, unido a los efectos de los altos niveles de estrógenos y progesterona sobre el metabolismo de las lipoproteínas78,79. En ausencia de estudios apropiados y en función de la experiencia actual, este Grupo de Consenso recomienda una discusión a fondo y una evaluación CV detallada en mujeres que deseen quedarse embarazadas. Si no está contraindicado el embarazo, se recomienda de forma clara mantener la aféresis de LDL.
Recomendaciones del Grupo de ConsensoLas recomendaciones de este Grupo que ha realizado la adaptación española del documento de Consenso Europeo de HFHo se resumen en el tabla 3. El diagnóstico precoz de la HFHo y el inicio inmediato de una terapia hipolipemiante son primordiales. Mientras que normalmente el análisis genético puede proporcionar un diagnóstico definitivo de HFHo, los médicos deberán ser conscientes de que en algunos pacientes la confirmación genética seguirá siendo difícil8. El nivel de cLDL no debe ser el único criterio para el diagnóstico, considerando las nuevas evidencias de que la heterogeneidad genética de la HFHo se traduce en variabilidad fenotípica a un nivel mayor que el que se pensó previamente.
El tratamiento actual de la HFHo debe centrarse en la intervención sobre el estilo de vida, junto con la terapia con estatinas potentes a dosis máximas, en combinación con ezetimiba (en algunos casos con otros tratamientos modificadores de lípidos) y aféresis de LDL (fig. 10), de acuerdo con las guías más recientes8,80. A pesar de esta multiplicidad de tratamientos, es bien sabido que la mayoría de los pacientes con HFHo no alcanzan los objetivos de cLDL recomendados y que, por tanto, siguen presentando un alto riesgo CV. La reciente aprobación de lomitapida como tratamiento complementario específico para la HFHo, junto con el potencial de la inmunoterapia con inhibidores de PCSK9, ofrece a estos pacientes la posibilidad de una reducción adicional del cLDL respecto a los tratamientos de referencia actuales. Considerando los beneficios frente a los riesgos y el coste, que pueden diferir de un país a otro, el uso de tales fármacos podría traducirse en una mejor evolución clínica de los pacientes con esta rara enfermedad genética y potencialmente mortal.
Financiación
Esta adaptación del sobre hipercolesterolemia familiar homocigota de la Sociedad Europea de Arteriosclerosis ha sido elaborada por un comité científico delegado de la Sociedad Española de Arteriosclerosis y la Fundación Hipercolesterolemia Familiar que ha trabajado de forma independiente sin interferencias externas hacia sus aportaciones. Praxis Pharmaceutical ha colaborado en los gastos derivados de las reuniones de trabajo y el apoyo logístico sin intervenir en los contenidos del documento.
Conflictos de interesesLas siguientes declaraciones son sobre honorarios por conferencias/comités de asesoramiento, consultoría, ayuda para viajes y/o becas de investigación. Juan F. Ascaso: honorarios por conferencias y participación en comités de científicos de Amgen, Sanofi, MSD, Danone, Esteve, Recordati, Ferrer, AstraZeneca, Novartis, Lilly y Praxis Pharmaceutical; Pedro Mata: honorarios por conferencias y/o comités de asesoramiento de AstraZeneca, MSD, Amgen y Sanofi; Cristina Arbona: ha realizado colaboraciones con Fresenius Medical Care; Fernando Civeira: honorarios por conferencias y/o comités de asesoramiento de MSD, Ferrer, AstraZeneca, Esteve, Amgen, Sanofi, Pfizer y Synageva; Pedro Valdivielso: honorarios por conferencias y comités de asesoramiento de MSD, Ferrer y Chiesi; Luis Masana: honorarios por conferencias y comités de asesoramiento de Amgen, Sanofi, MSD, Kowa, Danone, Esteve, Recordati y AstraZeneca.
Metodología de trabajoEl Comité de Redacción se reunió 2 veces: una por teleconferencia y otra en Madrid, en reuniones presididas por el Dr. Luis Masana. En la primera reunión se expusieron todos los cambios propuestos por los miembros del comité y se aprobaron las primeras adaptaciones del documento. En la segunda reunión se validaron los cambios definitivos. Todos los miembros del Panel hicieron sugerencias de adaptación del documento original y revisaron el borrador completo. Todos los miembros del Panel estuvieron de acuerdo en la concepción y el diseño, participaron en la interpretación de los datos disponibles y sugirieron revisiones. Todos los miembros del Panel aprobaron el documento final antes de su envío a publicación.