




As is widely known, the classic function of HDL is reverse cholesterol transport (RCT), thus removing cholesterol from peripheral tissues. Early epidemiological studies, such as Framingham's, stated that increased HDL levels were associated with a significant decrease in relative risk for cardiovascular disease (CVD) mortality. However, those with heightened expectations in recent years for the development of therapeutic targets to increase HDL levels have been disappointed, because efforts have demonstrated the opposite effect on cardiovascular and global mortality.
However, in contrast, studies have highlighted the complexity and the intriguing role of HDL in different pathological conditions, such as infections, neoplasms, and autoimmune diseases.
In this review an attempt is made to summarize some biological pathways that link HDL function with the immune system, and its possible clinical repercussions in autoimmune diseases.
La función clásica de las partículas de colesterol HDL en el transporte reverso de colesterol está ampliamente establecida. Estudios epidemiológicos clásicos, tales como Framingham ya establecieron la relación inversa entre el incremento de los niveles de HDL y la mortalidad por riesgo cardiovascular.
Las grandes expectativas para el desarrollo de terapias que incrementen los niveles de colesterol HDL han creado grandes decepciones en estudios relativamente recientes. A pesar de todo, estos estudios han destacado la complejidad de las partículas HDL en diferentes condiciones patológicas como infecciones, neoplasias y enfermedades autoinmunes.
En esta revisión intentamos resumir algunos mecanismos biológicos que unen las HDL con las funciones dentro del sistema inmune y sus posibles repercusiones clínicas en las enfermedades autoinmunes.
As is widely known, the classic function of HDL is reverse cholesterol transport (RCT), removing cholesterol from peripheral tissues. Early epidemiological studies, such as Framingham's, stated that increased HDL levels were associated with a significant decrease in relative risk for cardiovascular disease (CVD) mortality.1 However, those with heightened expectations in recent years for the development of therapeutic targets to increase HDL levels have been disappointed because efforts have demonstrated the opposite effect on cardiovascular and global mortality.2
In contrast, however, studies have highlighted the complexity and the intriguing role of HDL in different pathologic conditions, such as infections, neoplasms and autoimmune diseases. These heterogenic functions of HDL have not been well understood, but what appears to be clear is that not only HDL-c levels but also lipoprotein particle composition appear to be important for the function of this complex lipoprotein.3,4
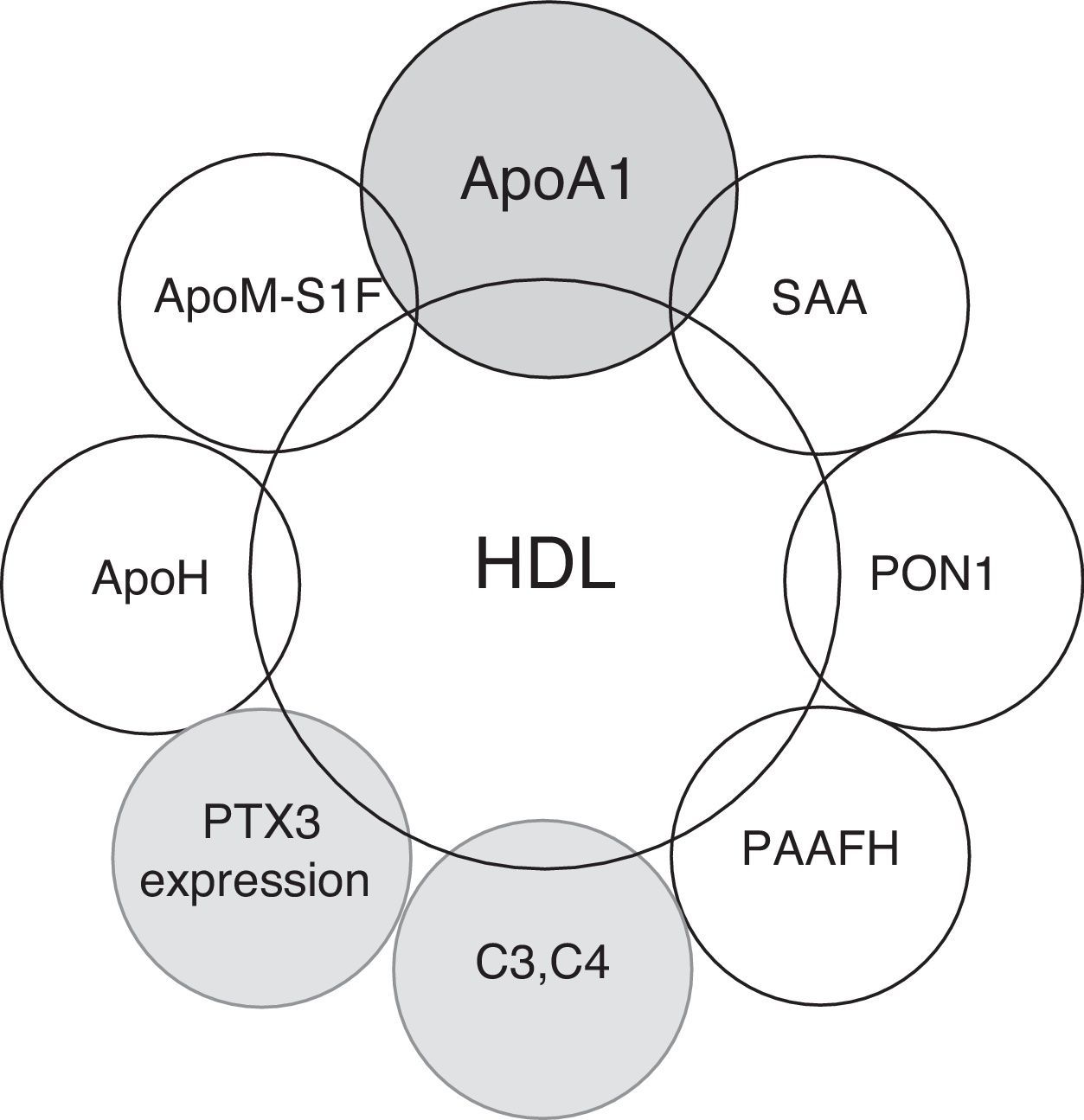
The predominant lipoprotein content in the plasma of several species is HDL. The main apolipoprotein associated with HDL is Apolipoprotein A1 (ApoA1), which is associated with cholesterol transport in cell surfaces via ABCA1 and ABCG1.5 ApoA1's structure is conserved throughout its evolution, and recent studies have associated HDL function not only with the homeostasis of cholesterol metabolism but also with immune system regulation,6 the acute phase response after infections; environmental stresses, such as severe burns; autoimmune diseases; and cancer.7–11
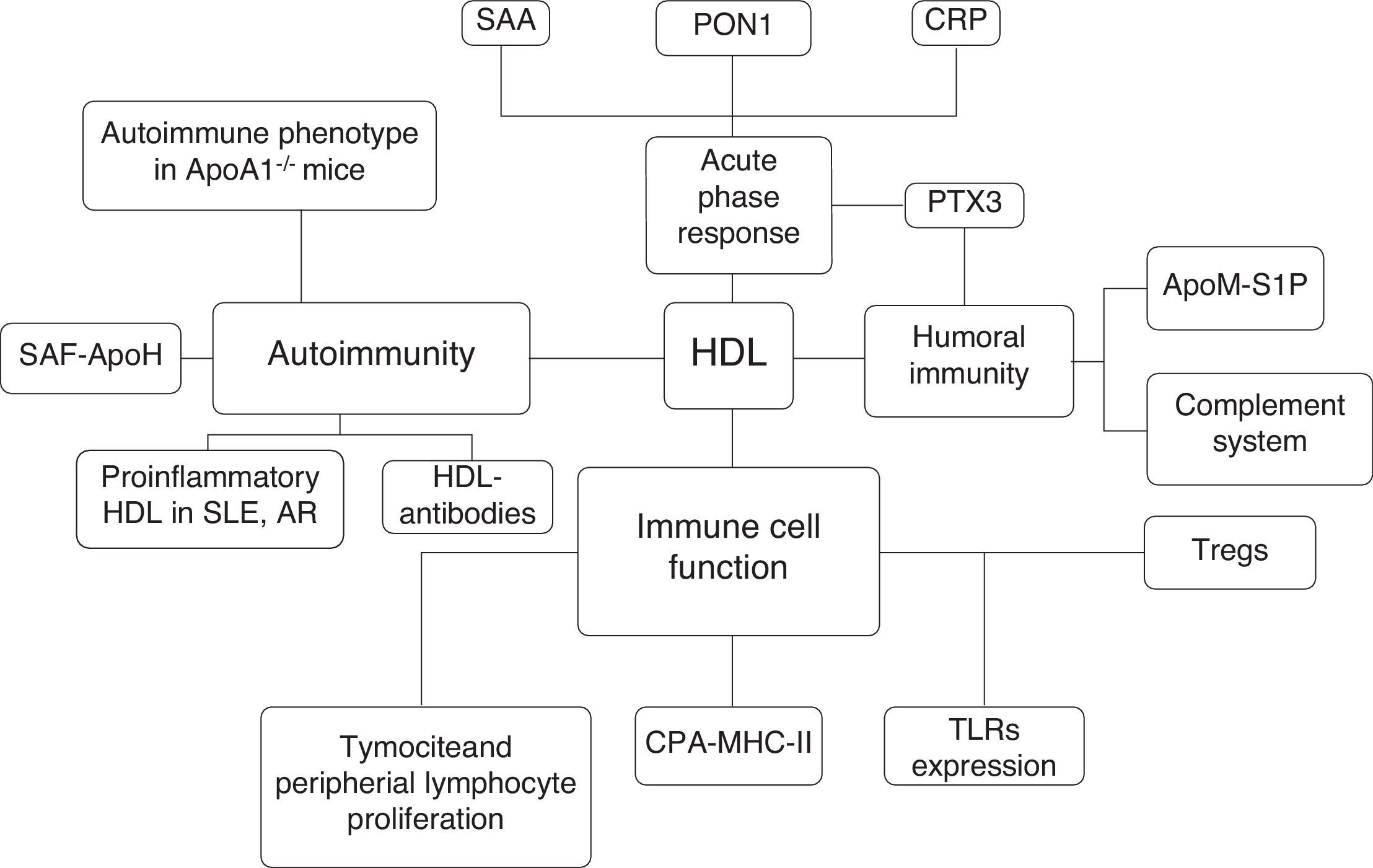
The proper knowledge of the role of HDL particles in pathologic conditions other than atherosclerosis has increased interest in the development of new therapeutic strategies for clinical conditions beyond cardiovascular diseases.4,12 In this review, we attempt to summarize the possible clinical relevance of HDL's functioning in the immune system in relation to its possible clinical implications in autoimmune diseases (Fig. 1).
HDL and the acute phase response
The potential protective nature of HDL has been primarily attributed to its role in RCT. However, the mechanisms by which HDL may impact cardiovascular health and disease remain complex and not fully understood.3,4 HDL possesses a number of heterogenic functions that impact cardiovascular health. The heterogenic functions of HDL involve anti-inflammatory, antioxidant, antithrombotic, antiapoptotic and nitric oxide synthesis mechanisms. These different functions are related to the complex and heterogenic structure of HDL particles. This heterogeneity is the result of changes in the content of the apolipoproteins, lipids and proteins that are associated with HDL and are related not only to cholesterol metabolism but also to regulating the complement system and the acute phase response.13–17
The acute phase response (APR) is a systemic reaction to infectious and noninfectious tissue destruction. Multiple physiologic adaptations occur, including changes in the hepatic synthesis of a number of plasma proteins, termed acute-phase reactants.18 Two acute-phase reactants, C-reactive protein (CRP) and serum amyloid A protein (SAA), are known to interact with lipoproteins.19,20 CRP binds to apolipoprotein B, which is contained in atherogenic lipoproteins, whereas SAA circulates primarily with HDL.
Apolipoprotein A1(ApoA1) and enzymes associated with HDL with antioxidant properties such as paraoxonase-1 (PON1), platelet-activating factor acetylhydrolase (PAF-AH), lecithin: cholesterol acyltransferase (LCAT) and glutathione selenoperoxidase (GSPx) are replaced by SAA after the acute phase response.21–28
Under these pro-inflammatory conditions, the composition of HDL particles changes; they evolve into pro-inflammatory, pro-atherogenic particles that have been associated with the presence of coronary artery disease (CAD) and an increased risk for cardiovascular diseases.17,21,29–35
HDL and immune cell functionLipid rafts and immune cell functionCholesterol membrane homeostasis is linked to the innate and adaptive immune response. Lipid rafts are membrane micro-domains that are enriched in cholesterol, phospholipids and proteins. They play an important role in activating signaling pathways in immune cells.6,36–38
HDL particles and cholesterol efflux transport mediated by the ATP-binding cassette A1 (ABCA1) or ABCG1 alter the structure and lipid composition of the cell membranes.39 Cholesterol depletion of lipid rafts by ApoA1 in antigen-presenting cells (APC) inhibits dendritic cells differentiation and the ability of macrophages and dendritic cells to stimulate T-cell activation by reducing the number of major histocompatibility (MHC) class II molecules.40,41
In another noteworthy study, it was demonstrated that ABCG1 negatively regulates thymocyte and peripheral lymphocyte proliferation, another novel mechanism by which cholesterol can alter the signaling pathways and proliferation of immune cells. These results showed that ABCG1 is a negative regulator of lymphocyte proliferation.42
It can be hypothesized that HDL may inhibit antigen presentation to T-cells by reducing the lipid raft cholesterol.7
Disruption of cholesterol efflux via ApoA-I can be used as a basis for interpreting the effects on immune cell function. One notable study demonstrated for the first time that cholesterol enrichment cells stimulate T-cell activation and expansion, developing an “autoimmune phenotype” that was resolved by subcutaneous injection of lipid-free ApoA1. The study's authors experimented with mice that lacked both LDLr (LDLr−/−) and apoA1 (LDLr−/−, apoA-I−/−) mice. As expected, when the LDLr−/− and apoA-I−/− (DKO) mice were fed with an atherogenic diet, they developed increased atherosclerosis compared with the LDLr−/− (SKO) controls that were fed the same diet. Unexpectedly, the mice also displayed an unusual expansion and activation of the T-cells in their skin, which drained the lymph nodes and led to an autoimmune phenotype. Furthermore, when the T- and B-cells were fluorescence-activated and cell sorted from the lymph nodes of the atherogenic-diet-fed DKO mice, the cells were found to be nearly filled with CE, as measured by mass spectrometry, whereas the cells from the atherogenic-diet-fed SKO mice were not.43,44
Cholesterol enrichers other than macrophages and monocytes induce cell activation and cell dysfunction, developing autoimmune phenotypes.38
In addition to enriching the cholesterol in lipid rafts, it was demonstrated in another study that the decreased cholesterol in these micro-domains from neutrophiles by HDL downregulated the activation, adhesion, spread and migration of the neutrophiles.45,46
All of these data indicate that the RCT and HDL regulation of cholesterol homeostasis was not primarily intended for atheroprotective functionality. It is reasonable to consider HDL lipoproteins as part of the innate immune system that regulates the host response after infections. However, cholesterol's enriching of membrane micro-domains is associated with a stimulation of the immune system and possibly with a risk of autoimmune diseases and atherosclerosis.7,47
Activation of toll-like receptors mediated by HDL particlesToll-like receptors (TLRs) are molecules that are expressed in the monocytes, macrophages and dendritic cells (DC) involved in the innate immune response against infections. They are responsible for recognizing the conserved molecular patterns present in pathogens. In atherosclerotic lesions, TLRs play a role in recruiting leukocytes and in forming foam cells by activating T-lymphocytes. HDL interacts with TLR4 and inhibits the antiviral response in macrophages induced by lipopolysaccharides (LPS). ApoA1 and cholesterol removal from lipid rafts by ApoA-1 and the apoA1 mimetic peptide 4F also reduce TLR4 expression.48–51
Moreover, although direct evidence for any HDL effects on B-cell functioning has not yet been demonstrated, it seems possible that removing cholesterol from lipid rafts could also affect B-cell activation via the expression of BLR.7,52,53
T regulatory cell (Treg-CD4+CD25+FoxP3+)Regulatory T-cells (Tregs) are the guardians of peripheral tolerance, acting to prevent autoimmune diseases, such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). Defects in Tregs have been reported in these two diseases despite significant differences in their clinical phenotypes and pathogeneses. In both diseases, the potency of the Tregs fails to keep pace with the activation of effector cells, and the Tregs are unable to resist the ensuing inflammation.54,55
The importance of Tregs is underscored by the overwhelming inflammation and autoimmunity that result from their absence. Treg cells have been linked not only to autoimmune diseases but also to the atherosclerotic process by inhibiting pro-inflammatory T-cells in atherosclerotic vessels.56–58
Of note, in an autoimmune animal model in mice, apoA1 treatment increased Tregs and decreased the percentage of effector/effector memory T-cells (Teff), suggesting that apoA1 may control T-cell homeostasis in peripheral lymphoid tissues by regulating the Treg/Teff balance. How apoA1 modulates Treg/Teff balance remains unknown, but the process likely involves cellular cholesterol and lipid raft homeostasis.43,59,60
HDL and humoral immunityComplement system and HDLA number of complement system proteins, activation products, receptors and regulatory proteins have been detected in atherosclerotic lesions, and C5b-9 deposits have been shown to correlate with the atherosclerotic process.61–63
Furthermore, complement component 3 (C3) plasma levels have been related to coronary artery disease measured by coronary angiography and to clinical ischemic events.64,65
Lipid and glucose metabolism are also related to the complement system. A number of studies have associated C3 and C4 levels with higher levels of triglycerides and postprandial hypertriglyceridemia, low levels of HDL and insulin resistance, which are the metabolic syndrome compounds.66
These data suggest the relevance of the complement system in atherogenesis.67
Furthermore, a fragment of C3 (C3a-des-Arg) is common to a portion of the acylation-stimulating protein (ASP), which is the most potent stimulant of triglyceride synthesis and glucose membrane transport in human adipocytes.68 This could help to explain the association between the complement system, lipid metabolism, insulin resistance and postprandial lipemia.69
More recently, it has been stated that HDL plasma levels interact with complement system activation because an inverse correlation has been identified between HDL level and the circulating terminal complex C5b-C9 levels70.
Recently, new proteomic technologies applied to HDL composition have revealed that HDL plays a previously unsuspected role in regulating the complement system. The complement components C3, C4 and C9, and complement-regulatory proteins, such as vitronectin and clusterin, have been found in HDL. These studies also showed that the HDL in patients with CAD was enriched with C3 and C4.14,15,23
Our group described in a previous work that in a cohort of patients affected with SLE, there was a positive correlation between small, dense HDL particles, C3 and C4 levels and the activation of the complement cascade.71 These small HDL particles were pro-atherogenic, and they were also associated with the presence of subclinical atherosclerosis and inflammatory serum markers such as ESR and CRP. These data highlight that in SLE patients, different HDL subpopulations with different compositions have different roles in autoimmune systemic disease, but there is a close association with the complement system. These small HDL particles were pro-atherogenic and pro-inflammatory, but the positive correlation with the complement system components may be important in the clinical course of lupus because a decrease in the complement proteins is associated with flare-ups and worse clinical courses.
PentraxinsPentraxins are a superfamily of acute-phase proteins that are highly conserved during evolution and characterized by the presence of a multimeric structural motif, the pentraxin motif. They can be classified as short pentraxins, such as C reactive protein (CRP), or long pentraxins, such as PTX3. PTX3 was the first recognized member of this family. It is produced in a number of cell types (mononuclear phagocytes, dendritic cells, fibroblasts and endothelial cells) in response to interleukin (IL)-1 and tumor necrotic factor (TNF), in contrast with CRP, which is produced only in the liver and is stimulated by IL-6.72 It would appear that PTX3 plays a role in the innate immune response given that it has been reported that PTX3 deficiency leads to invasive pulmonary aspergillosis owing to the defective recognition of conidia by alveolar macrophages and dendritic cells.73,74
PTX3 has also been associated with some types of cardiovascular disease, including atherosclerosis.75–77
Norata et al. demonstrated in a noteworthy study that HDL induces mRNA expression and the protein release of PTX3.78 Moreover, PTX3-C1q binding promotes complement-mediated clearance of apoptotic cells, limiting inflammatory tissue damage after injury. These data suggest that part of the atheroprotective effects of HDL could result from the modulation of molecules that act as sensors of the immunoinflammatory balance in the vascular wall, but no additional studies have confirmed this atheroprotective association with PTX3.72
Sphingosine 1-phosphate (S1P)Sphingosine 1-phosphate (S1P) is a membrane-derived lysophospholipid that acts primarily as an extracellular signaling molecule.79 Recent evidence on the physiology of the S1P-S1PR axis in the homeostasis of immune-mediated cells is receiving increased attention because these cells have become attractive therapeutic targets in diseases such as chronic inflammatory pathologies (asthma, arthritis), autoimmune disorders (multiple sclerosis), neoplasms and atherosclerosis.
Signals initiated by S1P are conducted by five G protein-coupled receptors (GPCR), named S1P15.79 Cellular and temporal expression of the S1P receptors determine their specific roles in various organ systems, but they are particularly critical for regulating the cardiovascular, immune, and nervous systems, with the most well-known contributions of S1PR signaling being modulation of vascular barrier function, vascular tone and regulation of lymphocyte trafficking.80
Approximately 35% of plasma S1P is bound to albumin and 65% to ApoM, which is found on a small percentage (approximately 5%) of high-density lipoprotein (HDL) particles.80 This ApoM+HDL-bound1P has been proposed as a primary contributor to the antiatherogenic properties of HDL.81
There are a number of well-characterized S1PR agonists and antagonists; however, most compounds have been directed toward modulating S1P1 activity. FTY720 (fingolimod; Gilenya, Novartis) is the prototypical S1PR agonist and was approved by the U.S. Food and Drug Administration as a first-line oral therapy for relapsing-remitting multiple sclerosis (MS).82–84 FTY720 acts as an agonist on S1P1 and S1P3–5, and it also acts as a functional S1P1 antagonist by inducing receptor endocytosis and degradation of this receptor.85–88 This promiscuity may be responsible for the adverse effects, such as bradycardia and hypertension, seen in fingolimod-treated patients.89
S1P receptors regulate many aspects of immune cell biology. The best known is S1P1's regulation of lymphocyte migration out of the secondary lymphoid organs into the blood and lymph nodes.
The contribution of S1PRs to regulating the immune response has been studied extensively in the context of experimental autoimmune encephalomyelitis (EAE), the most commonly used animal model of multiple sclerosis (MS).90 The mechanism of action of the S1P1 agonist fingolimod has been presumed to be the trapping of autoreactive T- and B-cells in the lymphoid organs, away from the central nervous system.91 However, T-cell S1P1 may also regulate the activation and differentiation of these immune cells. Deletion of T-cell S1P1 significantly suppresses the ability of these cells to be polarized to Th17 in vitro.92
S1P1 is also expressed on CD4 T-cells that have been isolated from human rheumatoid arthritis (RA) patients.93 S1P enhances the expression by these cells of the receptor activator of nuclear factor kB (RANK) ligand, an effect that is replicated in a synovial cell-like cell line. In collagen-induced RA models, an S1P1-specific antagonist prevented or ameliorated disease by up-regulating lymphocyte CD69 expression, which downregulates S1P1 surface expression, blocking thymic egress.93–95
S1P1 suppresses Treg development via the AKT/mTOR pathway and affects their migration from the thymus and out of the periphery by counteracting CCR7 retention signals, similar to the mechanism that regulates the egress of effector T-cells from lymph nodes.90,96,97 FTY720 significantly increased the number of regulatory T-cells while decreasing central memory T-cells.98
In LDL-receptor-deficient mice, fingolimod inhibited atherosclerosis by modulating lymphocyte and macrophage function; in this study, lipids were unchanged, but the lymphocyte blood count decreased.99
Although S1P1 has been the focus of much research, little is known of the roles of the S1PR with a cell-subtype-specific effect on certain cell lines in the immune system.
Research focusing on knowledge of the diverse biological functions of the S1PR family will provide an opportunity to discover new treatment strategies for these autoimmune and chronic inflammatory disorders. The study of the function of S1P that is transported by apoM-HDL is also of interest in inflammatory response and vascular permeability after infection. In one transversal study, decreased ApoM levels were associated with the presence of sepsis and systemic inflammatory response syndrome (SIRS) after infection.100
With all of these data, the HDL-apoM-S1P axis is of interest as a novel therapeutic approach with diverse biological functions in different pathogenic processes that involve the immune system.
HDL and autoinmune diseasesPlasma HDL-c levels are modified by a number of clinical conditions, such as autoimmune diseases. HDL-c is elevated in multiple sclerosis and reduced in systemic lupus erythematosus (SLE), rheumatoid arthritis (RA),68 Sjögren's syndrome,69 ankylosing spondylitis,70 psoriatic arthritis, and inflammatory bowel disease.71
Patients affected with chronic autoimmune diseases also maintain a chronic inflammatory status that appears to accelerate atherosclerosis and cardiovascular diseases.
Research on nonclassical cardiovascular risk factors and the interaction of these diseases with lipid metabolism is of interest because these diseases are characterized by chronic inflammation and hyperactive immune systems.101,102
HDL and SLESLE is the prototype of autoimmune diseases with multisystem involvement. The presence of antibodies with different targets is associated with the diversity of clinical manifestations of this systemic disease.
Patients affected with systemic lupus erythematosus (SLE) show an increase in cardiovascular mortality and morbidity despite improvements in the control of disease activity and complications.103,104 These data are supported by the results of a number of studies that have shown a higher prevalence of subclinical atherosclerosis in this young population.105–107 The accelerated atherosclerosis observed in patients with SLE cannot be entirely explained by the traditional cardiovascular risk factors. The presence of some metabolic disturbances, such as atherogenic dyslipidemia9 and metabolic syndrome (MetS), appears to be more prevalent in SLE patients owing to inflammatory mechanisms.108,109 Other nonclassical cardiovascular risk factors related to inflammation that have been associated with this accelerated atherosclerotic process include C-reactive protein (CRP), cytokines, the complement system110–112 and some adipokines.113,114
HDL metabolism is affected by the presence of lupus. A “lupus pattern” of dyslipidemia has been defined by elevated levels of very-low-density lipoprotein cholesterol (VLDL) and triglycerides (TG) and lower high-density lipoprotein cholesterol (HDL) levels.9,115 A notable finding has been that activity aggravates these alterations – striking increases in VLDL and TG levels and decreased HDL levels were directly correlated with SLEDAI scores.115 Another consideration regarding the lipid profiles of SLE patients is that ordinal biochemical analyses may not reflect changes in lipoprotein subpopulations that could give additional information about SLE's pro-atherogenic lipid profile, which has been described in studies on lipid profiles measured by magnetic nuclear resonance (MNR) and the presence of subclinical atherosclerosis.112
The abnormalities observed in SLE that are associated with low HDL-cholesterol levels could be explained by a number of mechanisms that involve inactivating lipoprotein lipase activity and the presence of antibodies against LPL and ApoA1. OxLDL/β2GPI complexes and IgG anti-β2GPI antibodies were also found in lupus patients with high TG and low HDL levels.116
As was described above, these HDL particles also show qualitative changes in composition and protein cargo, with HDL lipoproteins evolving into pro-inflammatory molecules.35,117,118
These “pro-inflammatory HDLs” were associated with the presence of subclinical atherosclerosis in SLE patients and also in RA patients.21
Changes in HDL composition after the acute phase response and these chronic inflammatory conditions impair the functionality and antioxidant properties of HDL.
One of the most important antioxidant enzymes bound to HDL is PON1. Decreased PON1 serum enzyme activity has been reported in SLE119,120 and also in other rheumatic disorders such as RA121 EM122 and psoriasis.123 The impaired antioxidant ability of HDL has been associated with increased atherosclerosis and also with the degree of disease activity.
Titers of antibodies against HDL and ApoA1 were associated in SLE patients with persistent inflammatory disease (measured with the SLEDAI index),124,125 and they have also been inversely correlated with paraoxonase activity.126
Investigation of HDL's impaired cholesterol transport capacity in autoimmune disorders is of interest owing to HDL's association with modulating the immune system. In SLE and RA patients, impaired HDL function as measured by efflux cholesterol capability has been described.127 Cholesterol efflux capacity was associated with the degree of disease activity in RA127 and with the extent of psoriatic areas and the severity index in another study.128 In this study, it was found that SLE patients showed a more complex pattern of modifications, with marked reduction of ATP-binding cassette G1- and ATP-binding cassette A1-mediated cholesterol efflux capacity unrelated to the disease activity.127
In another study with SLE patients, antibodies against ATP-binding cassette transporter A1 were associated with the presence of atherosclerosis, but it was not the study's objective to investigate ABCA1's association with the disease activity.
Therapeutic strategies using ApoA and ApoA-mimetic peptides as targets have been initiated in animal models of SLE.
As we described earlier, an animal model of apoA-I−/−LDLr−/− mice displayed an autoimmune-like phenotype similar to SLE, including increased plasma antibodies against double-stranded DNA, β2-glycoprotein I, and oxidized LDL. Treatment of these mice with lipid-free apoA-I reversed the autoimmune phenotype and lowered the number of lymphatic nodules.44,129
Another study, with a murine lupus model that was treated with apolipoprotein A-1 mimetic peptide (L-4F) both alone and with pravastatine, showed decreased levels of IgG anti-dsDNA, IgG anti-oxPLs, proteinuria glomerulonephritis and osteopenia.130
Apolipoprotein H and antiphospholipid syndromeApolipoprotein H (Apo H), also known as beta2-glycoprotein, is a glycoprotein involved in clotting mechanisms and lipid pathways. ApoH, is the main autoantigen responsible for negatively charged antiphospholipid antibodies production in the antiphospholipid syndrome (SAF) a prothrombotic clinical condition in patients with autoimmune diseases as lupus but also in the general population.131
Polz and Kostner described for the first time the presence of a beta2-glycoprotein in human apolipoproteins, and it was designated with the name of ApoH. After that, it was described that only a small percentage of ApoH (4–13%) is associated with plasma lipoproteins, for this reason the plasma levels of apoH have also been known as beta2-glycoprotein (beta2-GPI).132,133
Patients affected by SAF and SLE are under an oxidative stress environment. This oxidative stress is one of the mechanisms that promote important structural changes in beta2-GPI protein. One of these changes is the formation of beta2-GPI-oxLDL complexes that are associated with arterial thrombosis. Antibodies against these complexes have been reported both in SLE patients and in diabetic patients and were associated with an increase of the cardiovascular risk.134
To conclude, the parallelism between autoimmune diseases and the atherosclerotic process focuses on the role of HDL in modulating cholesterol homeostasis and the pathogenesis of both diseases. This promising approach could highlight new therapeutic strategies to modulate the immune system and disease activity and to avoid the metabolic disturbances that are associated with increased cardiovascular risk factors and the atherosclerotic process in patients with increased morbidity and mortality (Fig. 2).

Interactions between HDL particles and the immune system. SAA: serum amyloid A; PON: paraoxonase-1; CRP: C-Reactive protein; PTX3: pentraxin 3; S1P: sphingosine 1 phosphate, apoM: apolipoprotein-M; Tregs: T regulatory cell; APC: antigen presentation cell; MHC-II: major histocompatibility complex II; SLE: Systemic lupus erythematosus; Rheumatoid Artritis, SAF: antiphospholipid syndrome; ApoA1: apolipoprotein-1.
The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of DataThe authors declare that no patient data appears in this article.
Right to privacy and informed consentThe authors declare that no patient data appears in this article.
Conflict of interestThe authors confirm that this article content has no conflict of interest.